遗传算法(GA)导论
本文最后更新于 2025年9月1日 上午
遗传算法导论
A genetic algorithm tutorial, Darrell Whitley, 1994
遗传算法的概念
遗传算法是一类将特定问题潜在的解决方案编码并组织到形如染色体(chromosome)结构的数据结构(下文直接称之为染色体)上,然后应用推荐算子(recommend operators)对数据结构中的特定信息进行保留的算法。
遗传算法的操作对象是一组这样的染色体,称之为种群(population)。遗传算法会针对这样的种群进行评价,然后筛选:对于目标问题有更优解的染色体会被赋予更高的机会进行复制和重组,称为”繁殖”(Reproduce)。
优化问题
遗传算法遇到的问题大多可以归结为两类:编码问题和评估函数的问题。
编码(Encoding)问题
对于参数的选取,通常认为参数之间应该具备线形不相关的特性,但事实上很难保证参数的这一性质,在遗传算法中,参数之间的关联称为基因关联(Epistasis)。
遗传算法对与参数的编码建立在一个基本的假设上:代表系统参数的变量可以用字节串(位串,Bit string)来进行表达,这也意味着这些变量能够以某种方式被离散化。离散化的范围以\(2^n\)表示,例如如果每个参数都用10bit来表示,那么经过离散化后,我们能至多得到1024个离散值(1024种01的组合)。对参数作离散化处理是为了在保证精度的前提下,给系统输出提供尽可能大的分辨率(Resolution)以便调整系统输出。
字节化编码的问题在于离散值的冗余,如果变量离散化后有1200个(介于1024和2048之间)离散值,那么需要用11个字节才能表示完全,但是如此会有2048-1200=848个无用的离散值,这些无用的离散值可能会导致没有评估,或者是出现不好的评估结果。
评估函数(Evaluation function)问题
评估函数能够对系统的输出进行评估。构建评估函数的过程是一个模拟(仿真)系统的过程,这样的仿真相比与真实系统必然只能给出近似解或者是部分解。因此评估函数对系统输出的结果只是近似的,或是部分的评价。
对于遗传算法来说评估函数的计算速度是一个问题:首先,对于现有种群的评估计算量就比较大。不仅如此,种群的后代也需要进行评估。
搜索空间(Search space)
假设参数之间的关系是非线形的,如果用于表示参数的比特数为L,那么参数空间的大小是\(2^L\),遗传算法在这样的一个\(L\)维的超空间(Hypercube)中采样。这个超空间的大小随着L指数型增长,将带来巨大的采样难度和计算量。
经典遗传算法
算法流程
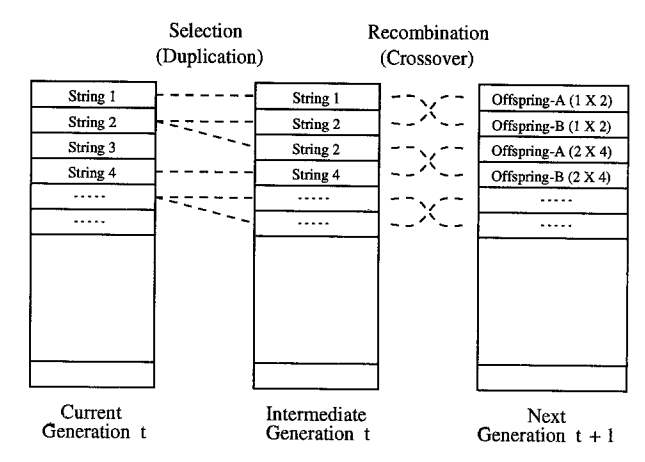
经典遗传算法通过对当前种群的评估(Evaluation),选择(Selection),重组(Recombination)和突变(Mutation) 后,能够在现有种群的基础上产生下一代种群。
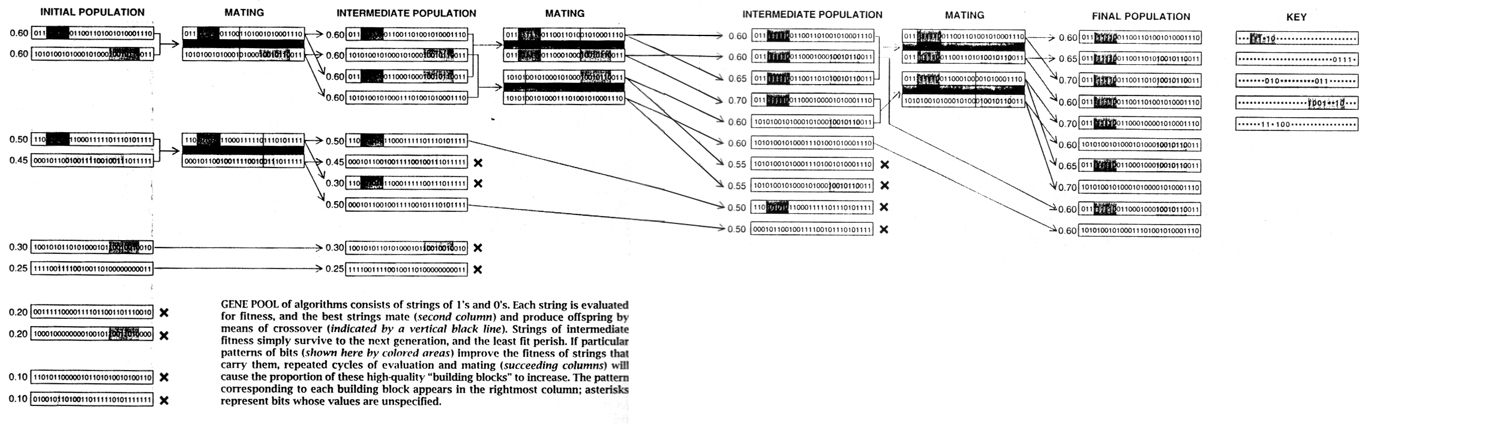
原始种群(Initial population)
在遗传算法中,定义种群中编码后的一条固定长度为L的字节串称为一个个体,这样的个体称为基因型(Genotype)或者是染色体(Chromosome)。 在大多数情况下,原始种群被随机的产生出来。
此处的随机指在空间内随机的取出一些编码的组合(当然也受制于编码机制等)。
评估和适应度
原始种群被生成后,每一个个体会通过评估函数和适应度函数(Fitness funtion)生成其对种群的适应度(Fitness)。
评估函数和适应度函数两个概念一般来说是可以替换的。事实上,评估函数是通过一组参数来实现对个体表现的衡量,而适应度函数是通过增殖(Reproductive)概率来衡量个体表现的。 评估函数的衡量不依赖于种群中的其他个体,适应度函数的衡量与种群中其他个体有关。
种群中个体\(i\)的适应度定义为:
\[\frac{f_i}{\overline{f}}\]
其中,\(f_i\)表示评估函数对第\(i\)个个体的评估结果,\(\overline{f}\)表示种群的平均评估。
选择
定义在现种群经过选择后保留的种群称为中间种(Imermediate population),中间种经过突变和重组后会成为下一代种群。
在遗传算法中,选择的本质是概率性地将现种群中的个体进行复制,最终得到中间种的过程。
选择机制有非常多种,其中最为常见的是轮盘赌(Roulette wheel)和余数随机采样(Reminder stochastic sampling).
- 轮盘赌
如果整个轮盘表示整个种群,每个个体所占面积与适应度成正比,每一次转动就能随机地抽取一个个体复制,更高效的办法是轮盘外围上均匀地分布着N个均等间距的指针,每一次转动就能随机抽取N个个体进行复制。 经过若干次转动后,结果的集合构成中间种。
- 余数随机采样
具体而言,对于适应度大于1的个体,适应度的整数部分表示该个体会被复制多少次。对所有的个体,适应度的小数部分表示额外被复制的概率。 比如,适应度2.3的个体能够获得2次复制,并且有0.3的概率能获得第三次复制的机会。
重组
- 单点交叉
遗传算法中重组的本质是杂交(Crossover),其过程主要有两步:- 随机地使得个体间两两配对。
- 随机地选取一对个体,两者在某个随机且相同的比特位处断开,前后的两段基因型进行交叉互换。
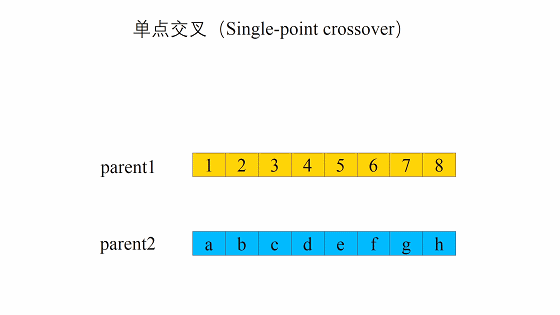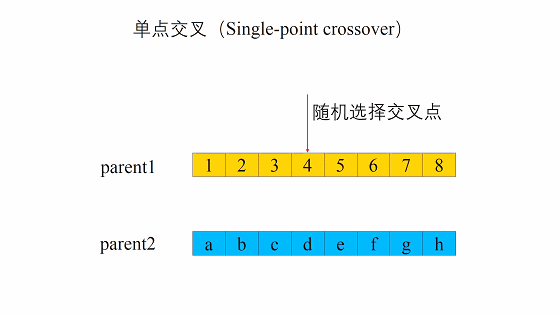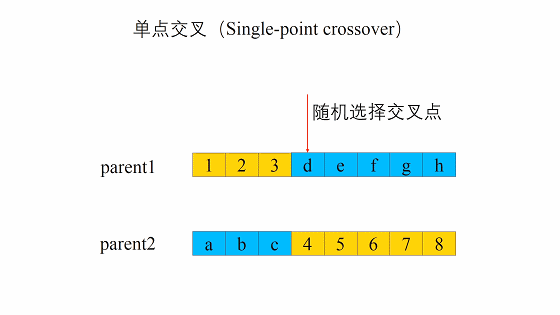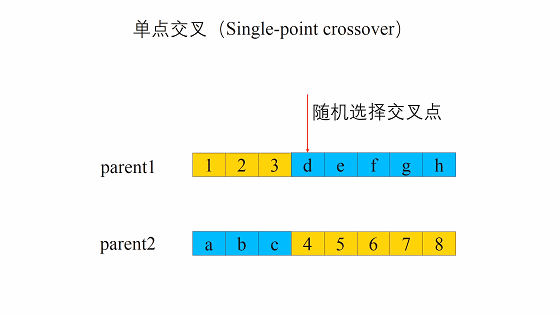
- 随机地使得个体间两两配对。
- 两点交叉
两点交叉是指在个体染色体中随机设置了两个交叉点,然后再进行部分基因交换。两点交叉的具体操作过程是:- 在相互配对的两个个体编码串中随机设置两个交叉点;
- 交换两个个体在所设定的两个交叉点之间的部分染色体。
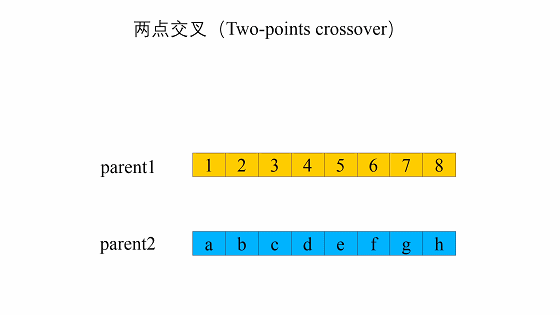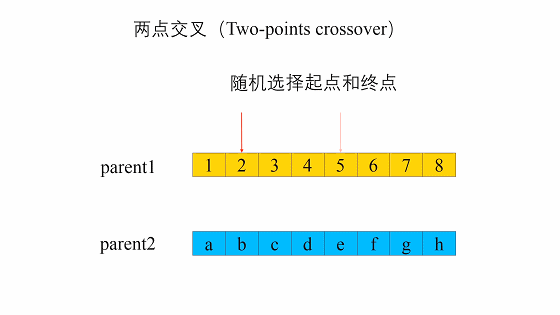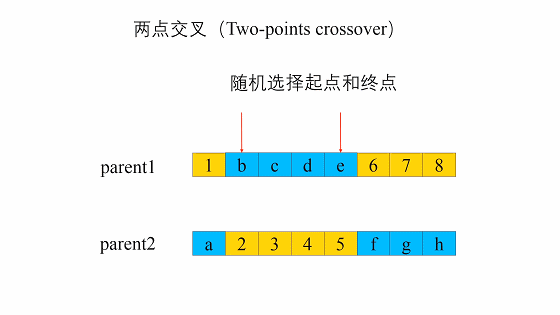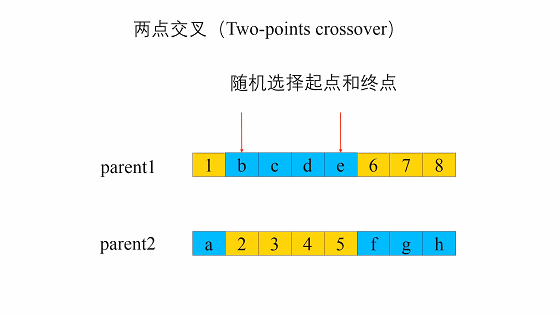
- 在相互配对的两个个体编码串中随机设置两个交叉点;
新生成的两个个体称为后代(Offspring),后代能够插入到下一代的概率计作\(p_c\)。
突变
重组之后利用突变算子对后代作突变处理,对于种群中的所有比特位,其突变的概率计作\(p_m\),这是一个非常小的值,通常小于1%。 突变有可能随机地产生一些比特(并且有50%的可能性改变原本的比特值),也有可能反转原有的比特(一定能改变原本的比特值)。
中间种经过重组和突变,最终能称为新的种群。
可行性分析
经典遗传算法的鲁棒性和复杂性建立在样本空间中有偏向性的超平面采样之上。
搜索空间和超平面
在几何学上,称n维空间的某一个小于n维的子空间为超平面(hyperplane), 比如二维空间的超平面是一条线,三位空间的超平面是一个面。
在位串长度固定位\(L\)的前提下,种群中所有可能的编码方式所构成的空间称为搜索空间(Search space)。 如果每一种特定的编码方式在L维搜索空间中对应了一个角(Corner),那么共超平面的几个角对应的编码中必定在相同的某几个比特位上的值是相同的,此时引入通配符(Don‘t care,以*记)的概念,那么搜索空间的一个超平面就可以表示为含有Don’t care(*)的位串(比如:0****,11*****),这样的位串称为模式(Schema),每一个模式对应了一个超平面。
复制:偏向性地采样
对于一个\(K\)维的搜索空间:***…***,该空间能够被分割为1**…***和0**…***两个超平面,对这两个超平面分别计算平均适应度,采样过程总是有高概率地采集适应度更高的超平面内的样本。在下一次采样中,这个超平面再度被等距划分为多个子部分,采样会更倾向于平均适应度更高的子部分,重复若干次采样后,采样总是更倾向于采集整个搜索空间中适应度最高的部分。因此通过这样的采样能够在搜索空间中找到适应度最高的部分。换句话说,遵循这样规律的复制总是能够有高概率地选择到种群中适应度更高的个体。
采样过程的本质是一个在搜索空间中不断寻找局部最大值的过程。
其次,定义选择后某一特定超平面\(H\)内留存的样本的期望数目为该超平面现有的样本数目\(M(H,t)\)与该超平面适应度均值\(f(H,t)\)的乘积: \[E=M(H,t) \times f(H,t)\] 这样的采样方法会使得采样后在特定超平面的样本数与其期望大致相符合,事实上经过选择后\(H\)留存的样本数目\(M(H,t+1)\)可以被公式化为:
\[M(H,t+1)=M(H,t)\frac{f(H,t)}{\overline{f}}\] 其中\(\overline{f}\)表示整个种群的平均适应度,近似为1。
重组:产生新的样本的同时带来破坏
由于复制不会产生新的样本,而选择可能会减少种群中的样本数目,为了避免最终种群的个体数目过小,同时产生新的可能性,因此在每一次采样后需要用染色体重组(交叉)来产生新的样本。 不仅如此,染色体重组(交叉)还可以起到部分地保留当前的采样超平面倾向的作用,下面来讨论染色体重组对原来染色体中信息的破坏(Disruption)程度:
对于一组一阶染色体,其染色体中的信息必不可能受到染色体重组的影响。
对于二阶和二阶以上的染色体组,其破坏程度和交叉点的数目有关:
如果只有一点发生染色体重组,染色体组在一点交叉后发生比特变换(破坏)的概率受Schema中确定字符(该位置上的值不是通配符)的位置决定。
如果有两点发生染色体重组,则在互补位置发生交叉时对染色体组的破坏最大。
总而言之,染色体中的原本信息在重组中受到破坏的程度与模式中确定字符的位置有着密切的关系。 不管是一点交叉还是两点交叉,可以发现Schema中相邻的既定比特位受到重组带来的破坏最小。
定义距
称在交叉过程中被整体保留下来的确定字符称为适应性等位基因(Coadapted alleles)。在某一个模式当中,两个确定字符之间的距离称为两个字符的联系(Linkage),第一个确定字符的位置和最后一个确定字符的位置之间(这个部分称为有义部分(Significant Portion of Schema))的距离称为模式的定义距(Defining length),以\(\Delta(H)\)记。 > 两点及两点以上的交叉中需要将Schema组织为首尾相连的环状形式,才能得出其定义距。
通过之前对重组破坏的推演可以得出结论:破坏程度与定义距的长度有关:定义距的长度越长,在交叉过程中原本染色体信息被破坏的可能性就越大。不仅如此,定义距直接反映了交叉发生在有义部分的概率,对于一点交叉而言,一点交叉发生在有义部分的概率为: \(\frac{\Delta(H)}{L-1}\)。
倒换
除了交叉和变异之外,遗传算法中还会用到的对染色体的基本操作是倒换(Inversion)。 倒换是随机的将染色体中的某一段序列进行镜像翻转。倒换可以改变染色体上确定字符之间的连接,这样具有更大非线性的确定字符在染色体上的间隔距离可能会被缩小。
倒换的前提是比特以一种位置无关的方式进行编码(否则倒换就等同于大规模变异)。一种可行的编码方式是染色体上的每一个基因以(位置,值)的形式表示。比如对于010010110而言,染色体为((9,0)(6,0)(2,1)(7,1)(5,1)(8,1)(3,0)(1,0)(4,0))。 对于倒换和如此表示所带来的问题,在此不做过多叙述。

模式定理
推导过程
模式定理(Schema theorem)提供了进化时采样率的下界,推导过程如下:
由之前提出的经过选择后超平面\(H\)留存的样本(个体)数目\(M(H,t+1)\):
\[M(H,t+1)=M(H,t)\frac{f(H,t)}{\overline{f}}\]
考虑重组对选择后超平面\(H\)中种群样本数目的影响:
- 重组是有概率发生的,概率为\(p_c\)。
- 对于发生重组的种群,交叉既有可能产生出现有空间内某个模式的副本(比如100和010交叉就可能产生000,使得000的副本增加一个),同时也有可能使得原有的样本消失。
那么现在后代中落在超平面\(H\)的样本数目:
\[M(H,t+1)=(1-p_c)M(H,t) \frac{f(H,t)}{\overline{f}} +p_c [M(H,t)\frac{f(H,t)}{\overline{f}}(1-losses)+gains]\] 为了简化计算,忽略gain,并且假设发生在Schema上有义部分的交叉必然导致染色体破坏,记破坏概率为\(disruption\),那么有:
\[M(H,t+1) \geq (1-p_c)M(H,t) \frac{f(H,t)}{\overline{f}} +p_c [M(H,t)\frac{f(H,t)}{\overline{f}}(1-disruption)]\] 定义超平面\(H\)的采样率表示超平面\(H\)的样本数目与种群中样本数目的比,以\(P(H,t)\)记。
由之前对定义距的理解,对原信息的破坏只可能发生在定义距的区间段内。此外,如果发生重组的位串都在平面\(H\)内,那么重组也不可能对原本信息造成破坏,因此要想让重组破坏原有的信息,亲本中的另一条位串必定来自于其他平面。
由这上述两点可以将破坏概率定义为:
\[\frac{\Delta(H)}{L-1}(1-P(H,t))\] 由破坏的定义可以得出如下结论:
定义距\(Δ(H)\)越小,模式受到破坏的概率就越小。从直观上来说,定义距越小,交叉发生在定义距内(即一定能破坏信息)的概率也越小。
那么下一代超平面\(H\)的采样率可以表示为: \[P(H,t+1) \geq P(H,t) \frac{f(H,t)}{\overline{f}} [1-p_c \frac{\Delta(H)}{L-1}(1-P(H,t))]\]
如果考虑亲代是基于适应度选择出来的:
\[P(H,t+1) \geq P(H,t) \frac{f(H,t)}{\overline{f}} [1-p_c \frac{\Delta(H)}{L-1}(1-P(H,t)\frac{f(H,t)}{\overline{f}})]\] 最后,考虑突变的影响:记突变发生的概率为\(p_m\),超平面\(H\)的阶数为\(o(H)\),那么表示超平面\(H\)的Schema不会受到突变影响的概率为:
\[(1-p_m)^{o(H)}\] 可以得出结论:
模式的阶数\(o(H)\)越小,模式不会受到突变影响的概率越大。从直观上来看,模式的阶数代表着有效字符的个数,有效字符越少,在交叉过程中越容易被保留下来。
最终,超平面\(H\)在下一代中被采样到的概率可以表示为:
\[P(H,t+1) \geq P(H,t) \frac{f(H,t)}{\overline{f}} [1-p_c \frac{\Delta(H)}{L-1}(1-P(H,t)\frac{f(H,t)}{\overline{f}})] (1-p_m)^{o(H)} \] 可以通过数学推算出,在适应度\(\frac{f(H,t)}{\overline{f}}>1\)时: \[P(H,t) \frac{f(H,t)}{\overline{f}} [1-p_c \frac{\Delta(H)}{L-1}(1-P(H,t)\frac{f(H,t)}{\overline{f}})]=P(H,t) \frac{f(H,t)}{\overline{f}}(1-p_c \frac{\Delta(H)}{L-1})+[P(H,t) \frac{f(H,t)}{\overline{f}} ]^2\] 用\(t=0\)代来推算\(t\)代时候的采样率:
\[P(H,t) ≥ \{P(H,0) \frac{f(H,0)}{\overline{f}}(1-p_c \frac{\Delta(H)}{L-1})+[P(H,0) \frac{f(H,0)}{\overline{f}} ]\}^t(1-p_m)^{o(H)}\] 可以发现:在适应度\(\frac{f(H,t)}{\overline{f}}>1\)时,采样率呈现指数型上升。
可以总结为:
在选择,重组,突变算子的作用下,当某个超平面的适应度大于1时,模式的阶数\(o(H)\)越小,定义距\(Δ(H)\)越小的个体越能够被保留下来,且数目成指数型上升。
模式定理在数学上证明了重组和突变的有效性,并给出了采样率的下界,是遗传算法中重要的理论基础之一。
突变和收敛问题
显然,模式定理最强调交叉和超平面采样在遗传搜索中的作用。为了在选择后最大限度地保存超平面样本,应尽量减少交叉和突变的破坏。这表明突变可能根本不应该使用,或者至少应该在非常低的水平上使用。
突变的积极作用是突变可以防止任何特定位点或等位基因的永久丢失(尤其是对种群中个体数目非常小的时候而言), 同时突变也增加了种群的基因多样性。
随着代数的增加,选择有可能使得在某些位置上的比特全部变成相同的值,表明算法收敛。但是与此同时,个体之间适应度的差别会越来越小,选择的影响也会越来越小,最终可能导致提前收敛。可以通过对适应度的缩放来改善这一问题。
关于算法的收敛:遗传算法的收敛性一直是一个问题,简单而言,当选择的代数达到一定程度后,选择压力减弱,导致种群中适应度优秀的个体数始终处于一个水平上而不发生变化,此时称遗传算法达到收敛。和深度学习不同的是,如果在收敛后继续学习,那么优秀个体的数目在理论上并不会下降。
重组的采样方式·均匀交叉
交叉点数目问题
在某个范围内,交叉点的数目多一些,破坏的影响随之减弱。但是过多的交叉点数目会导致出现非常大的破坏。
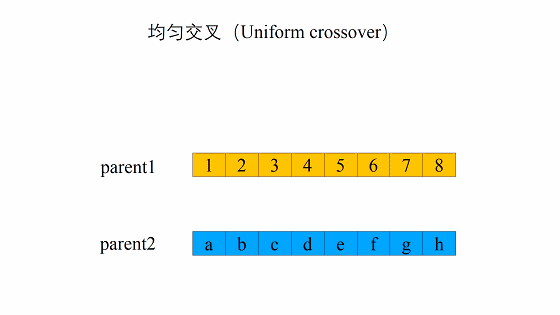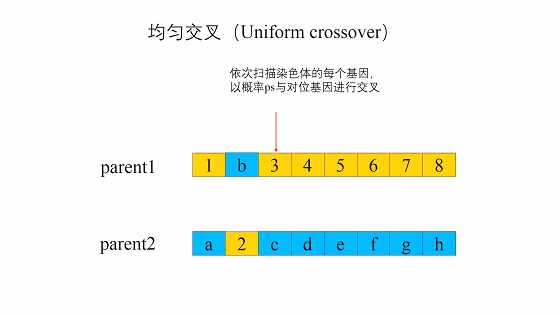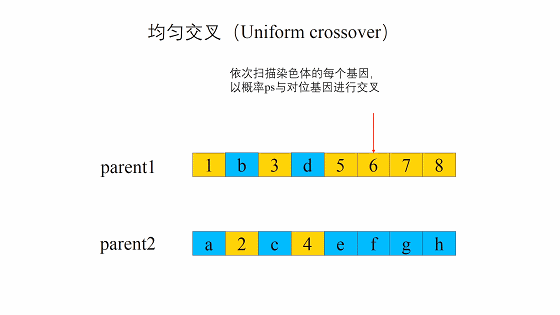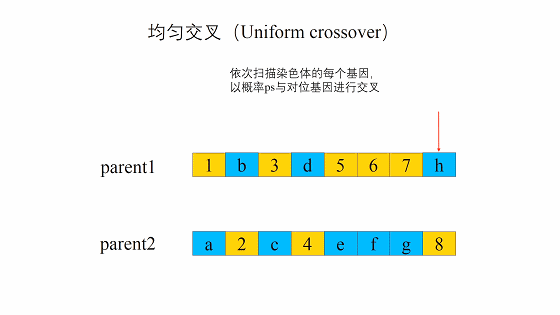
均匀交叉(Uniform crossover)
均匀交叉的流程是首先随机对种群中的染色体进行两两配对,和单点交叉不同的是,均匀交叉随机地选择亲代染色体中的某些位置,然后对换亲代染色体上这一位置上的比特值。 与单点交叉相比,均匀交叉的破坏概率不受定义距的影响,均匀交叉的破坏概率为:
\[1-(\frac{1}{2})^{o(H)-1}\] 虽然相比于单点交叉均匀交叉的破坏概率更大,但是对于个体数目小的种群,更大的破坏概率能解除信息量小的限制。
同时,在采样方式上,相比与单点交叉,理论上均匀交叉能够采集到更多的样本,具体的可由Fig 4 说明。 如果把两个位串中同一位置上的比特值不相同的位置数称为海明距离(Hamming distance),记作\(\mathcal{H}\),那么理论上均匀交叉可以产生\(2^\mathcal{H}-2\)种不同的位串,而单点交叉只能产生\(2(\mathcal {H}-1)\)种不同的位串。
简化取代表示
对于两个位串,将同一位置上比特值相同的地方用“-”表示,不同部分保留的表示方法称为简化取代表示(Reduced surrogates)法。 例如,对于0001111011010011和0001001010010,就可以表示为—-11—1—–1和—-00—0—–0,如图所示。
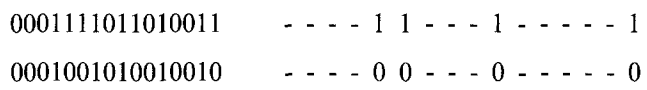
这样的表示法好处是可以方便的理解这两个位串的重组其实是发生在一个4维空间中的,由于均匀交叉的距离无关性,可以发现对亲代的破坏就发生在这些点上,因此可以将均匀交叉简化为在这种表示方法下的一点交叉。 其次,如果至少一个交叉点存在于这种表示方法的第一个确定字符和最后一个确定字符之间,那么能够保证交叉结果绝对不同于亲代,这意味着能够在这个空间内采到新的样本。
种群大小
低阶的超平面拥有比较高的采样率。一个采样空间可被划分为\(2^iC_L^i\)个\(i\)阶的超平面。超平面的数量与研究种群大小\(N\)和遗传算法能够采样到的超平面数有关。通常认为超平面的数量与一开始的随机种群中的个体数量是一个指数关系:在\(N\)大小的种群里,\(i\)阶的超平面大约会被采集到\((\frac{1}{2})^i N\)个样本。
\(N^3\)理论
\(N^3\)理论认为,当种群大小为\(N\)时,遗传算法处理的有效超平面个数为\(N^3\)。 记\(\theta\)为至少复制\(\phi\)次后种群中超平面的最高阶数,\(\theta=log_2(\frac{N}{\phi})\),那么有采样空间中\(\theta\)阶的超平面总数一定大于等于遗传算法处理的超平面个数:
\[2^\theta C_L^\theta \geq N^3\] 长度为\(L\)的模式总数为\(3^L\)。根据上述理论,如果选用\(N=3^L\),那么至多有\(N\)个超平面可以被遗传算法处理。 因此种群大小要根据\(L\)进行选择。
模式理论的局限性
- 忽略了gains和低估了losses的影响。
- 当搜索逐渐聚拢在一些特定的超平面的子空间内时,观察到的超平面\(H\)的适应度会有巨大变化。因此取平均适应度为分母的方法仅在前几代可行。
- 字符串采样的偏差使得用模式理论计算并预测结果并不可行。
总而言之,模式定理没有为遗传算法的行动轨迹提供确切的描述,也无法预测特定超平面是如何随着时间的推移而处理的。
具体的标准遗传算法模型
下述内容中提出了一种利用标准遗传算法思路的算法模型,这种算法模型解决模式理论的局限性,并对模式理论进行一些定量计算。
还原模式定理推导的第一步:
\[M(H,t+1)=(1-p_c)M(H,t) \frac{f(H,t)}{\overline{f}} +p_c [M(H,t)\frac{f(H,t)}{\overline{f}}(1-losses)+gains]\] 现在从个体的视角来看,设某一个位串\(Z\)在下一代中被留下的概率为\(P(Z,t+1)\),上述式子可以改写成:
\[P(Z,t+1)=P(Z,t)\frac{f(Z,t)}{\overline{f}}(1-\{p_c losses\})+\{p_c gains\}\] 如果将这个式子应用于搜索空间中的每一个个体,那么就能够将遗传算法进行定性的计算。
损失和增益
在交叉的过程中,损失来源于两个亲本在交叉后子代为现有种群中没有的新位串,(此时认为由于交叉后亲本没有被保留,因此原有的亲本信息受到了损失),而增益来源于两个亲本在交叉后产生的子代与现有种群中的另一亲本相同(相当于现有种群中的某一个位串被复制了一次)。
对于某一个个体而言,损失和增益都是可以被计算的,下述一例:
令\(Z=000\),其损失可以按照如下方式计算:
\[losses=P_{I0}\frac{f(111,t)}{\overline{f}}P(111,t)+P_{I0}\frac{f(101,t)}{\overline{f}}P(101,t)+P_{I1}\frac{f(110,t)}{\overline{f}}P(110,t)+P_{I2}\frac{f(011,t)}{\overline{f}}P(011,t)\]
\(P_{I0}\):表示在与000进行交叉时,任何一位中出现交叉的概率,\(P_{I0}=1\)
\(P_{Ii}\):第\(i\)位与第\(i+1\)位之间发生交叉的概率。
增益也可以用同样的方式进行计算。
这种计算方式可以由采样空间\(S\)中的每一个位串\(S_i\)与目标位串\(Z\)做异或运算求得。
> \(S_i\)表示\(S\)中的位串按照特定顺序进行排列后的第\(i\)个位串,未作特殊说明时,这个排列顺序为从0依次+1。即:\(S_0=0_{2(二进制)}\),\(S_i=i_2\)。
标准形式下单点交叉的损失
形如:0000000000与0010000100,如果两个位串\(B\),\(B'\)满足如下条件:
\[B:相同-b-相同-b-相同\] \[B':相同-b'-相同-b'-相同\] 相同:\(B\)和\(B'\)在这些位置上的比特是相同的。
如果\(Z\)与\(S_i\)满足这样的关系,那么在第二个相同部分发生的交叉必然会导致亲本信息的损失。因此\(Z\)与某个特定的\(S_i\)进行单点交叉发生损失概率可以写作: \[\frac{δ(S_i)}{L-1}\]
\(δ(S_i)\):表示\(Z\)与\(S_i\)的最大连续相同部分的比特数。
\(Z\)在搜索空间\(S\)中由单点交叉产生的损失(总概率)可以描述为:
\[losses=∑\frac{δ(S_i)}{L-1}\frac{f(S_i,t)}{\overline{f}}P(S_i,t)\]
单点交叉的增益
要想让两个位串通过交叉产生位串\(Z\),那么以位串中的某一个位置为断点,其中一个位串在这个断点之前的部分与\(Z\)完全相同,另一个位串在这个断点之后的部分与\(Z\)完全相同。
具体而言:如果两个位串\(S_{α+x}\)和\(S_{ω+y}\),其中\(S_{α+x}\)在第\(α-1\)位置之前与\(Z\)连续相同,\(S_{ω+y}\)在第\(L-ω\)位置之后与\(Z\)连续相同,记\(ρ(S_{α+x},S_{ω+y})\)表示\(S_{α+x}\)和\(S_{ω+y}\)重叠部分的长度,那么两个位串在重叠部分发生交叉则必然会生成\(Z\),因此这两个位串交叉产生\(Z\)的概率为:
\[\frac{ρ(S_{α+x},S_{ω+y})}{L-1}\]
所以\(Z\)在\(S\)中由单点交叉产生的增益(总概率)可以描述为:
\[gain=∑\frac{ρ(S_{α+x},S_{ω+y})}{L-1}\frac{f(S_{α+x},t)}{\overline{f}}P(S_{α+x},t)\frac{f(S_{ω+y},t)}{\overline{f}}P(S_{ω+y},t)\]
概率矩阵
Vose和Liepins将\(S\)中两个位串\(S_i\)与\(S_j\)生成\(S_0\)的概率进行了矩阵化,下面是其矩阵化的步骤:
设\(s^t\)为一个向量,这个向量表示第\(t\)代\(S\)空间中每一个位串被选择的概率,\(s_i^t\)表示第\(t\)代中位串\(S_i\)被选择的概率,通常认为这个概率与适应度和前一次选择的概率之积成正比:
\[s_i^t = kP(S_i,t)f(S_i),k>0\] 那么\(S_k\)被选择出来的概率的期望值可以用如下公式来表示:
\[E(p_k^{t+1})=∑s^t_is_j^tr_{i,j}(k)\] \(r_{i,j}(k)\):\(S_i\)与\(S_j\)交叉产生\(S_k\)的概率。
\(r_{i,j}(0)\)可以用概率矩阵\(M\)表示,其中行表示\(S_i\),列表示\(S_j\),\(m_{i,j}=r_{i,j}(0)\),即\(S_i\)与\(S_j\)产生\(S_0\)的概率。
\(M\)的特性
- \(M\)的第0行和第0列表示的概率都是损失发生的概率
- \(M\)除了\(m_{0,0}=1\)在对角线上的其它值都为0
- \(M\)是一个对称矩阵
向量化
\(s^{t+1}\)可以用\(M\)进行表示: \[E(s^{t+1})=s^TMs\] 将\(M\)的上三角部分全部归零,得到矩阵\(M'\),通过之前的分析,\(M'\)的第一列表示\(s_0\)的损失,而\(s_i=P(S_i,t)\frac{f(S_i,t)}{\overline{f}}\),有:
\[S^TM'(:,1)s_0=P(S_0,t)\frac{f(S_0,t)}{\overline{f}}(1-losses)\]
记向量\(\sigma\)有:
\[σ_j<s_0,...s_{2^L-1}^T>=<s_{j⊕0},...s_{j⊕2^L-1}^T>\] 记\(\mathcal{M}(s)\):
\[\mathcal{M}(s)=<(σ_0s)^TMσ_0s,...,(σ_{V-1}s)^TMσ_{V-1}s>^T\] 矩阵\(F\)为对角线是\(f(i)\)的对角矩阵,有:
\[s^{t+1}=kFM(s^t),k>0\]
其他进化模型
除了标准进化模型之外,还有一些其他的进化模型。大致可分为进化策略(Evolutionary Strategies,ES)和进化编程(Evolutionary Programming,EP)两种。
进化编程中每一个个体是一个有限状态机(Finit-state machine),在此不做过多叙述。
进化策略中细分为两种类型:\((μ+λ)-ES\)和\((μ,λ)-ES\)。
在\((μ+λ)-ES\)机制中,亲代\(μ\)产生后代\(λ\)后,种群还会对亲代和后代共同进行选择,选择其中表现出色的个体生成下一代。在这种选择机制下,亲代会被保留直到被比亲代表现更出色的个体替代。
在\((μ,λ)-ES\)机制中,后代被产生后就直接替代亲代,选择在后代中执行。这种进化机制在选择阶段与经典遗传算法近似。但是在重组阶段所采用的算子与经典遗传算法不同。
\((μ+λ)-ES\)机制相比于\((μ,λ)-ES\)机制,其被优化的后代数目一定是单调增加的。
Genitor 算法
Genitor算法是一种使用\((μ+λ)-ES\)机制的算法,其与经典遗传算法中的进化模型不同点有三处。
- 选择在亲代中执行,选择后的亲代产生的后代被立即投放到下一代种群中。
- 后代不会替代亲代,但是每一代中适应度最差的个体被直接移除以加强选择压力。
- 适应度函数通过排名算法(Ranking)而非比值来表现。排名也同样能够保持选择压力的有效性。
排名算法:
设三个个体的适应度评估为:\(h_1,h_2,h_3\).
首先对所有个体按照适应度从小到大排序,比如:\(h_2,h_1,h_3\);
按照上面的顺序重新赋予fitness,即\(f(h_2)=1,f(h_1)=2,f(h_3)=3\)
计算选择概率:\(p(h_2)=\frac{1}{1+2+3}=\frac{1}{6},p(h_1)=\frac{2}{6},p(h_3)=\frac{3}{6}\)
CHC 算法
CHC算法是另一种能够单调选择位串的算法,CHC指Cross-generational elitist selection, Heterogenous recombination, Cataclysmic mutation。
Genitor算法是一种使用\((μ+λ)-ES\)机制的算法,具体的执行过程为:
在重组之后,亲代和子代中最好的\(N\)个个体生成中间种群,由于这样的选择已经能够制造足够的压力,因此CHC直接采用随机选择的方式从这个中间种群中进行挑选。同时,选择后的中间种中,海明距离远的两个个体才被允许进行繁殖。
在突变阶段,除了选择出来的优秀个体外,其余所有的个体都要经历相当大的突变,再进行交叉。
Hybrid 算法
Hybrid算法在个体编码时直接用实数进行编码,而非二进制数。其次,每个个体都在进行局部爬山算法(Local hill-climbing)来改善自身,产生后代之后,后代做爬山算法。
Hybrid算法通过这种多点局部搜索的方式使得搜索变得高效,局部爬山算法能够帮助改善染色体,但是不会对后代有太大的变化。基于上述特性,Hybrid算法在优化问题中的表现比较出色。
局部爬山算法:从当前的节点开始,和周围的邻居节点的值进行比较。 如果当前节点是最大的,那么返回当前节点,作为最大值 ( 既山峰最高点 ) ;反之就用最高的邻居节点来,替换当前节点,从而实现向山峰的高处攀爬的目的。如此循环直到达到最高点。
并行遗传算法
使用锦标赛算法(Tournaments)对现有种群进行选择就可以实现经典遗传算法的并行化。
锦标赛算法:
- 确定每次选择的个体数量N。(二元锦标赛选择即选择2个个体)
- 从种群中随机选择N个个体(每个个体被选择的概率相同) ,根据每个个体的适应度值,选择其中适应度值最好的个体进入下一代种群。
- 重复步骤(2)多次(重复次数为种群的大小),直到新的种群规模达到原来的种群规模。
锦标赛算法相当于是有噪声的排序算法。
具体的实现方法有两种模型:岛模型(island,图左)和细胞模型(cellular,图右)。
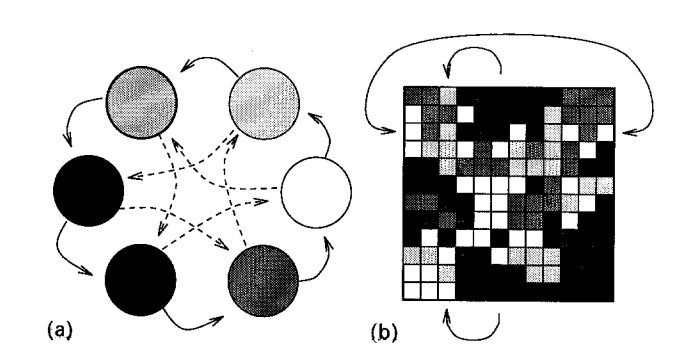
岛模型
岛模型将一个大种群均分为多个小种群,称为亚种(Sub-population),每个小种群中进行选择,并且每隔几代就将小种群中的一部分个体与另外的小种群中的个体进行交换,这个过程称为迁徙(migration)。
迁徙的目的是为了让小种群之间能够部分地交换基因信息。通过迁徙,岛模型更能挖掘每一个亚种内部的信息差异。
细胞模型
细胞模型将若干个简单处理元(processor)放在网格中,每一个处理单元处理一个位串,并且选择其相邻的单元中最优的位串,或者以一定概率选择相邻的某个位串与其进行配对,并且产生新的位串。 两个距离远的位串是无法进行配对的,这样的设定模拟了生物学上的地理隔离。 在几代之后,网格中会出现许多适应度接近的团块,随着进化的推进和选择的压力,这些团块的规模会随着适应度变大或者变小。