针对基因表达数据的聚类方法
本文最后更新于 2025年6月4日 晚上
针对基因表达数据的聚类方法
Cluster Analysis for Gene Expression Data: A Survey. Daxin Jiang et al, 2004.
DNA微阵列(基因芯片)简介:https://zhuanlan.zhihu.com/p/46017763
基因表达数据:https://baike.baidu.com/item/%E5%9F%BA%E5%9B%A0%E8%A1%A8%E8%BE%BE%E6%95%B0%E6%8D%AE/347523
SOM(自组织映射神经网络)——理论篇:https://zhuanlan.zhihu.com/p/73534694
背景
DNA微阵列技术
随着人类基因组(测序)计划(Human genome project)的逐步实施以及分子生物学相关学科的迅猛发展,越来越多的动植物、微生物基因组序列得以测定,基因序列数据正在以前所未有的速度迅速增长,传统的基因检测方式效率低下,为此,建立新型杂交和测序方法以对大量的遗传信息进行高效、快速的检测、分析显得格外重要。
cDNA微阵列技术/基因芯片技术(cDNA Microarrays/gene chip)是解决这一问题的重要技术:通过微加工技术将数以万计、乃至百万计的特定序列的DNA片段(基因探针),有规律地排列固定于2平方厘米左右的硅片、玻片等支持物上,构成的一个二维DNA探针阵列,其结构与计算机的电子芯片十分相似,所以被称为基因芯片。基因芯片主要用于基因检测工作。
基因表达数据
通过cDNA技术检测出来的基因数据称为基因表达数据(gene expression data)。基因表达数据反映的是直接或间接测量得到的基因转录产物mRNA在细胞中的丰度,这些数据可以用于分析哪些基因的表达发生了改变,基因之间有何相关性,在不同条件下基因的活动是如何受影响的。它们在医学临床诊断、药物疗效判断、揭示疾病发生机制等方面有重要的应用。随着 cDNA 微阵列和寡核苷酸芯片等高通量检测技术的发展,生物学家可以从全基因组水平定量或定性检测基因转录产物 mRNA。由于生物体中的细胞种类繁多,同时基因表达具有时空特异性,因此,基因表达数据与基因组数据相比,要更为复杂,数据量更大,数据的增长速度更快。
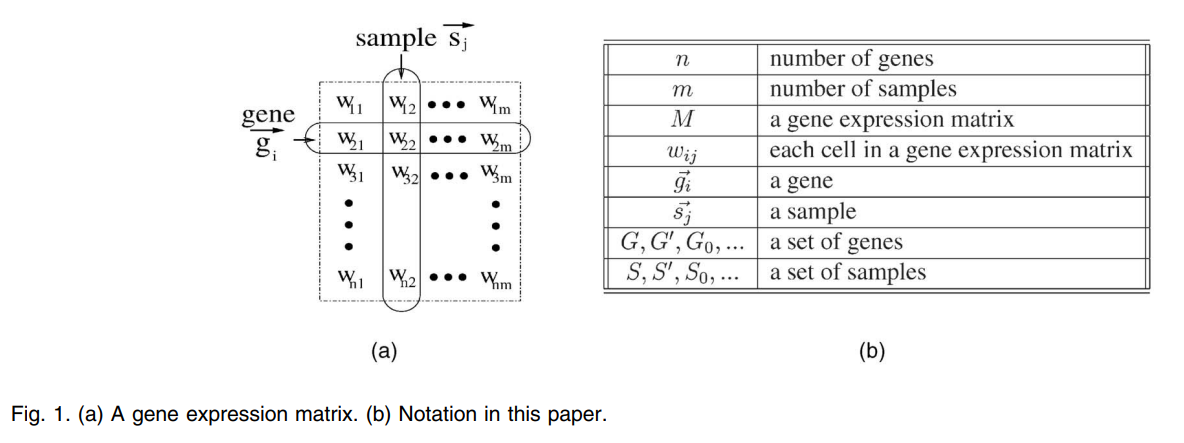
在本论文中,基因表达数据以矩阵的形式出现:矩阵的每一行\(\boldsymbol{g_i}\)表示每一个基因,矩阵的每一列\(\boldsymbol{s_j}\)表示一次在某一特定条件下对基因的表达实验,称为一次或者是一个样本(sample)。矩阵的每一个元素\(w_{i,j}\)表示基因\(g_i\)在实验\(s_j\)下的表达水平(expression level),表达水平是一个实数值。基因表达数据的组织形式如下图所示:
数据特征
- 矩阵的维度是及其不对等的,通常在微矩阵实验中的基因数量级在\(10^3\)到\(10^4\)左右,甚至可以达到\(10^6\),而参与的样本个数通常小于100,矩阵的长宽数量级差别非常大。
- 有些基因可能参与的共调控过程不止一个,因此个体之间的相关性高,聚类结果中的簇之间可能相互重叠。
- 通常,对于某个表现型,其参与该过程的基因数量占总参与实验的基因个数比例非常小,因此对于该表现型,无关基因(噪声)的数量非常大,信噪比非常低。只有一小部分基因参与任何感兴趣的细胞过程,而一个细胞过程只发生在一部分样本中。
数据处理
在实际的实验中,原始的矩阵中往往包含大量的噪声和缺失数据,因此需要对原始矩阵进行数据处理,常见的数据处理方式包括数据归一化和对缺失数据的预测。经过这些数据处理过程后才能探究基因表达相关的性质,本论文中假设这个原始矩阵已经被正确地数据处理过。
聚类方法
聚类是对基因表达进行分析的重要手段。聚类可以发现具有相同表达模式(expression pattern)的基因,称为共表达基因(coexpress gene)。从生物学的角度来说,共表达基因极有可能参与同一细胞过程,表示了基因的共调控机制(coregulation),并且为新的关于转录调控网络机制的假说提供参考依据。此外,聚类也可能发现新的一簇共表达基因类型。
机器学习中的聚类方法可以大致分为两类:监督的(supervised)和非监督的(unsupervised),二者的区别在于前者依赖于训练和提前指定好的类别,而后者仅根据数据自身的特征、不依赖于任何预先指定的数据类别进行分类。
在对基因表达数据的研究中,无论是对基因进行聚类还是样本进行聚类都是有意义的:对基因进行聚类可以找出共表达基因簇;对样本进行聚类,每个样本簇可能对应于一些特定的宏观表现型(例如在多个不同的样本中,实验条件中都有同一种癌症),比如癌症类型或临床综合征。
因此,本论文中按照对基因还是对样本分类将各种常见的聚类方法分类为三种:
- 基于基因的聚类(gene-based clustering):在这样的聚类中,基因被视为个体,样本被视为基因的特征。
- 基于样本的聚类(sample-based clustering):在这样的聚类中,样本被视为个体,基因被视为样本的特征。
有一些算法可以同时运用于这两种聚类。
- 子空间聚类(subspace clustering):在这样的聚类中,基因和样本被同等地视为个体和特征。
相似度度量
相似度(proximity)表示了两个数据的相似程度,在采样空间中表示为两个数据点的空间距离:两个数据点距离越近,两个数据点越相似。在基因表达数据中相似度度量有两种:欧氏距离(Eculidean distance)和皮尔森相关系数(Pearson correlation coeffitient)。
欧氏距离
欧氏距离是最常见的一种距离度量,其为高维空间中两点\(O_i=(o_{i1},...,o_{ip}),O_j=(o_{j1},...,o_{jp})\)之间的距离公式:
\[Eculidean(O_i,O_j)=\sqrt{\sum_{d=1}^p(o_{id}-o_{jd})^2}\] 如果某些维度上的数值数量级可能过大也可能过小,因此有必要在各维度上进行归一化处理。此外,经验证明欧氏距离对标准化\(O'_{id}=\frac{O_{id}-μ_{O_i}}{σ_{O_i}}\)后的基因表达数据的相似度度量要比非标准化的数据好得多。
皮尔森相关系数
另一种方法是皮尔森系数,皮尔森系数通过数据的统计特征来衡量两个数据之间的相似度,高维空间中两点\(O_i=(o_{i1},...,o_{ip}),O_j=(o_{j1},...,o_{jp})\)的皮尔森相关系数表示为:
\[Pearson(O_i,O_j)=ρ_{ij}=\frac{\sum_{d=1}^p(o_{id}-μ_{o_i})(o_{jd}-μ_{o_j})}{\sqrt{\sum_{d=1}^p(o_{id}-μ_{o_i})^2}\sqrt{\sum_{d=1}^p(o_{jd}-μ_{o_j})^2}}\] 其中\(μ_{o_i}\)和\(μ_{o_j}\)分别为\(O_i\)和\(O_j\)的均值。
由于皮尔森相关系数只根据数据的统计特性(均值和均方值)来衡量相似度,因此极有可能出现两个数据分布完全不同,但是在近似的地方出现最值被皮尔森相关系数判定为相似的情况。
改进这个问题的方法是刀切法(Jackknife),刀切法的相关系数表示为:
\[Jackknife(O_i,O_j)=min\{ρ_{ij}^{(1)},ρ_{ij}^{(2)},...,ρ_{ij}^{(l)}\}\] 其中\(ρ_{ij}^{(l)}\)指\(O_i,O_j\)都去掉第\(l\)个特征后的皮尔森相关系数。
从定义式中不难发现刀切法相比于皮尔森相关系数更为客观,其考虑到了每一个特征对总体分布的贡献。但是当特征数较大时,需要计算(特征数)次皮尔森相关系数,计算量较大。
此外,皮尔森相关系数假设了所有的数据服从高斯分布,对于实际上不符合高斯分布的数据,皮尔森公式的效果比较差。
改进这个问题的方法是 斯皮尔曼等级相关系数(Spearsman’s rank order correlation efficient)。它被定义成等级变量之间的皮尔森相关系数:对于样本容量为\(n\)的样本,\(n\)个原始数据被转换成等级数据,相关系数\(ρ\)为原始数据依据其在总体数据中平均的降序位置。斯皮尔曼等级相关系数虽然避免了皮尔森相关系数中对于数据分布的假设,但是使用等级来替代原有的数据使得大量的数据特征被丢失。
标准化的数据
前文中提到,如果对基因表达数据进行标准化处理,那么欧氏距离的度量效果会好得多。数据的标准化定义为:
\[O'_{id}=\frac{O_{id}-μ_{O_i}}{σ_{O_i}}\] 通过数学公式可以证明如下式子成立:
\[Pearson(O_i,O_j)=Pearson(O'_i,O'_j)\] \[Euclidean(O'_i,O'_j)=\sqrt{2p}(\sqrt{1-Pearson(O'_i,O'_j)})\] 上述式子可以说明,在标准化的前提下,皮尔森相关系数和欧氏距离的效果是相同的。
基于基因的聚类
K均值算法
K均值算法(K-means)是一种最基本的基于划分的(partition-based)聚类方法。在运行K-means之前需要设置类别数\(K\),K-means会最终将数据分为\(K\)类。
对于如图所示的数据集,使用K均值算法将其分成两类数据。 K均值算法的第一步是在数据集中随机生成两点,称为聚类中心/质心(Cluster Centroid)。(要分为多少类,就要生成多少个聚类中心) K均值算法是一个迭代算法,每一次迭代过程分为两部分:
- 簇分配
K均值算法会遍历每一个数据,计算该点与每一个聚类中心的距离,该点会被归属到最近的质心。
- 移动聚类中心
移动聚类中心到当前集群的平均位置
之后K均值算法会重复上述两步,直到聚类中心不再移动,此时可以认为聚类已经实现。
K均值算法的优化函数为:
\[E=∑_{i=1}^K∑_{O∈C_i}|O-μ_i|^2\] 其中\(O\)为\(C_i\)簇中的一个数据点,\(μ_i\)为\(C_i\)簇的质心。
因此K均值算法尝试最小化距离的各数据点到簇的质心的距离的平方和。
评价
K均值算法快而简单,往往经过很少的迭代就能够达到收敛。
K均值算法的缺点是,对于基因表达数据,往往\(K\)是不能通过先验知识可以确定的,因此需要使用不同的\(K\)多次执行K均值算法以找到最合适的\(K\)。这种做法对数据量大和潜在类别数多的数据集来说往往是不切实际的。
再者,K均值算法强制为每一个基因都会找到一个簇,即使这个基因可能并不适合每一个簇。对于信噪比非常低的基因表达数据,这些噪声也会被聚类到某些簇中,因此K均值算法对噪声非常敏感。
某些改进后的K均值算法通过设置某些全局参数来对K均值算法的运行过程进行限制,但是这些全局参数的设置仍然需要多次尝试以找到合适的设置。
自组织映射神经网络
自组织映射神经网络(SOM,Self-organizing map)是一种只有两层的无监督的神经网络。通过学习输入空间中的数据,生成一个低维、离散的映射(Map),从某种程度上也可看成一种降维算法。不同于一般神经网络基于损失函数的反向传播来训练,它运用竞争学习(competitive learning)策略,依靠神经元之间互相竞争逐步优化网络。且使用近邻关系函数(neighborhood function)来维持输入空间的拓扑结构。
SOM的结构如下:
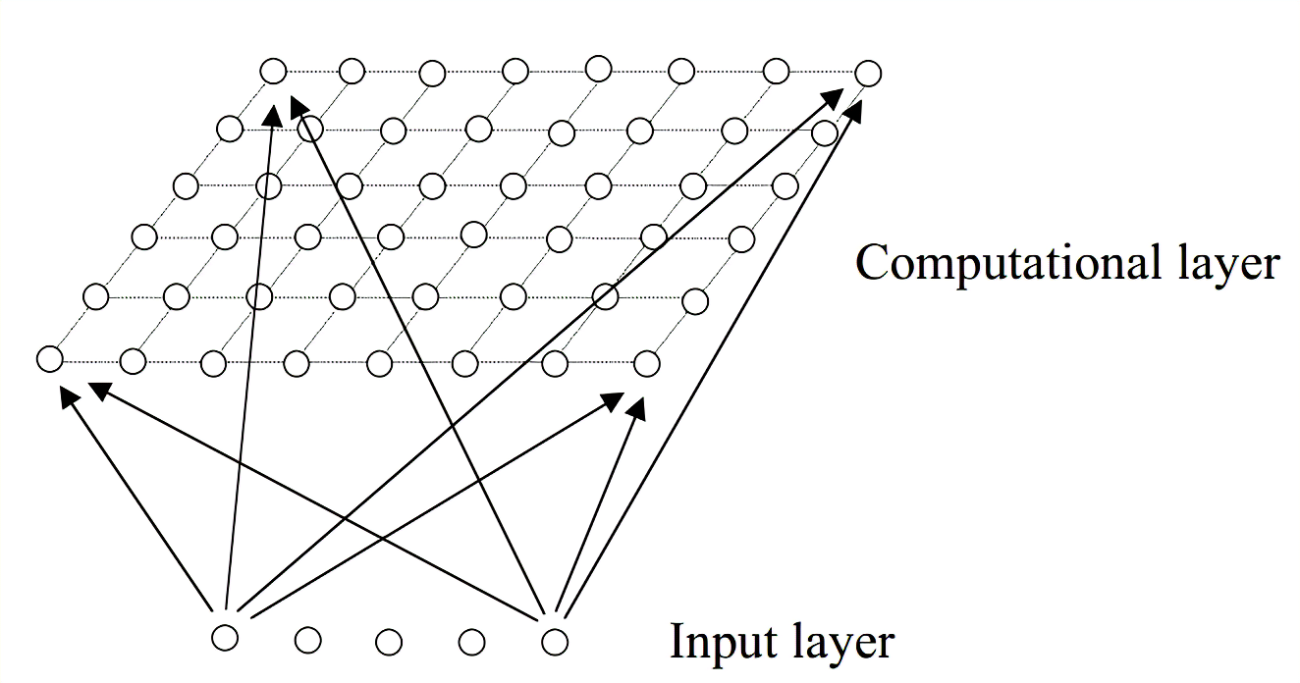
SOM只有两层,第一层为输入层,第二层为输出层,也称为竞争层(computational layer)。
输入层神经元的数量是由输入向量的维度决定的,一个神经元对应一个特征。
神经网络的每个神经元都与一个参考向量相关,每个数据点都被映射到具有 “最接近”参考向量的神经元上。在运行该算法的过程中,每个数据对象作为一个训练样本,引导参考向量向输入向量空间的密集区域移动,从而使这些参考向量被训练成适合输入数据集的分布。
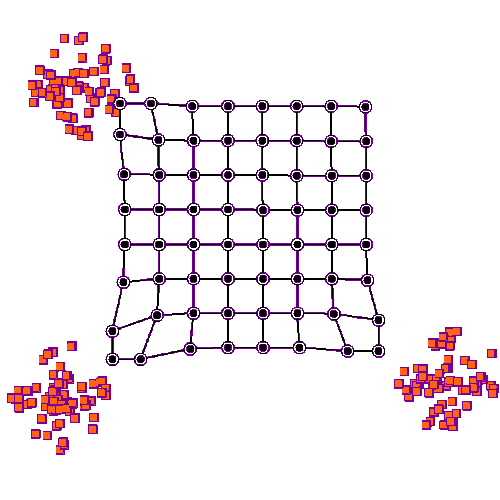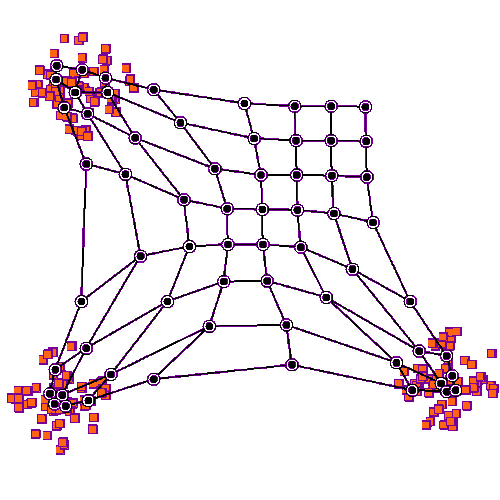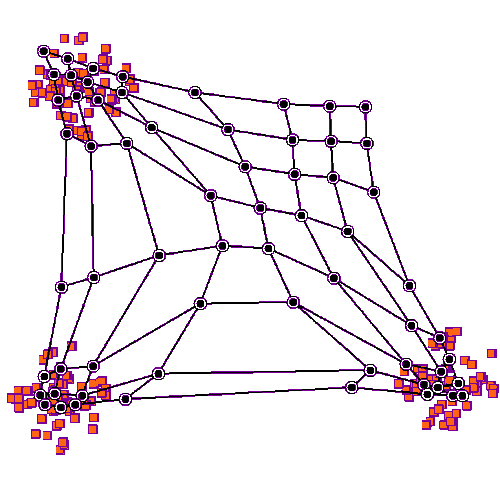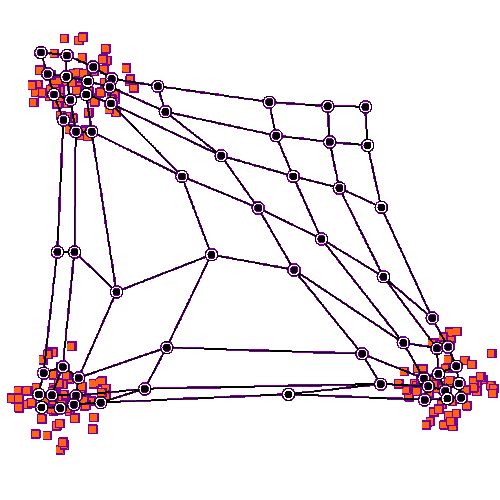
训练完成后,所有的数据点都被映射到竞争层的神经元,在竞争层观察到竞争层的神经元移动到输入层数据密集的区域。
评价
自组织映射神经网络最突出的特点是在输出层可以非常直观地观察到数据分布的特性;并且相比于K均值算法的抗噪性更好。但是自组织神经网络需要设定类别和初始竞争层的结构(如下图所示),这些参数的设置是一门学问。
如果数据集中有大量的与目标无关紧要的数据,在算法运行结束后,每一个簇中充斥着大量的这样的无关紧要的数据。
分层聚类
K均值算法和自组织映射神经网络都是基于划分的聚类方法,另一种聚类方法是分层聚类(hierarchical clustering)。与将数据直接划分为类别不同的是,分层聚类的聚类结果是相互交织叠加的类别,这些类别具有层级之分,层级可以通过聚类树状图(bendgram)进行表示,如下图所示:
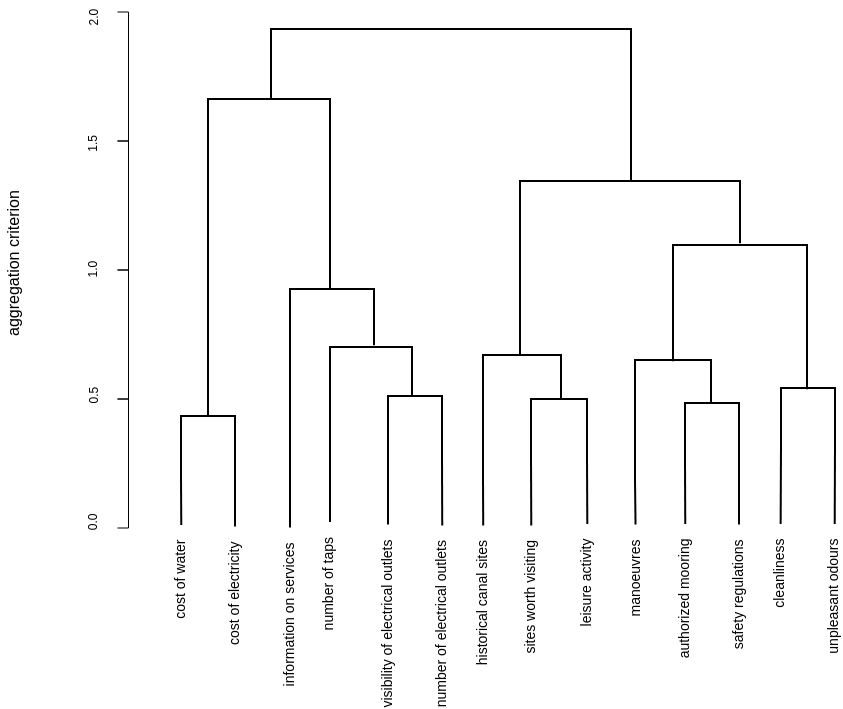
聚类树状图表示了类别之间的从属关系,可以允许根据需要的聚类层级来让用户自由选择聚类的程度。
分层聚类可以分为自上而下(divisive)和自下而上(agglomerative)两种算法类别。
自下而上的分层聚类最初将每一个数据点视为单独的一类,接下来的每一次迭代都根据度量将两个距离差最小的数据点为一类,最终所有数据全部聚为一类时,算法收敛。自下而上的分层聚类算法的度量通常是相似度(proximity)。常见的自下而上的分层聚类比如单链接聚类(single link)、完全链接聚类(complete link)、最小方差聚类等算法(minimum-variance)。
自上而下的分层最初将整个数据集中的所有数据视为一类,然后使用启发式算法或者基本图论对类别逐一划分,最终每一个数据点都为单独的一类时,算法收敛。
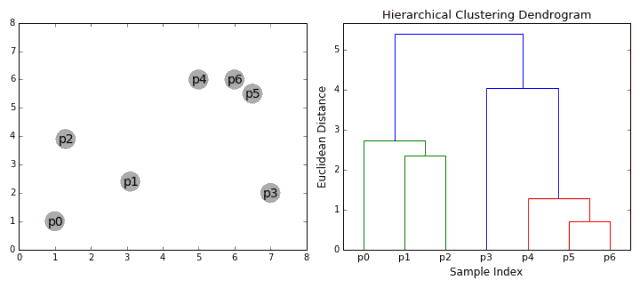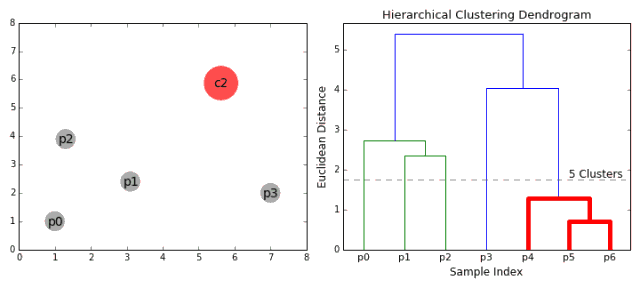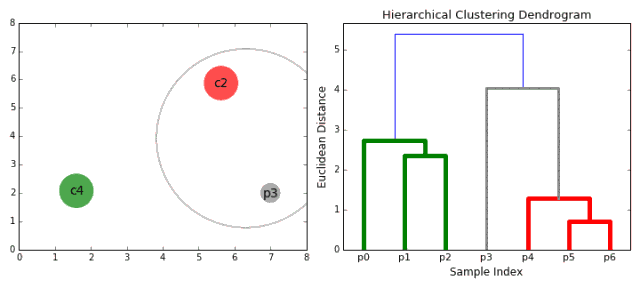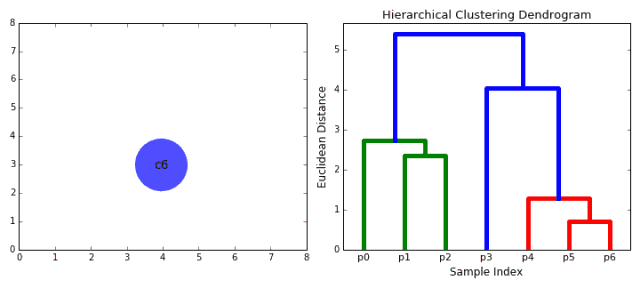
下图展示了一个对基因表达数据层次聚类的例子:
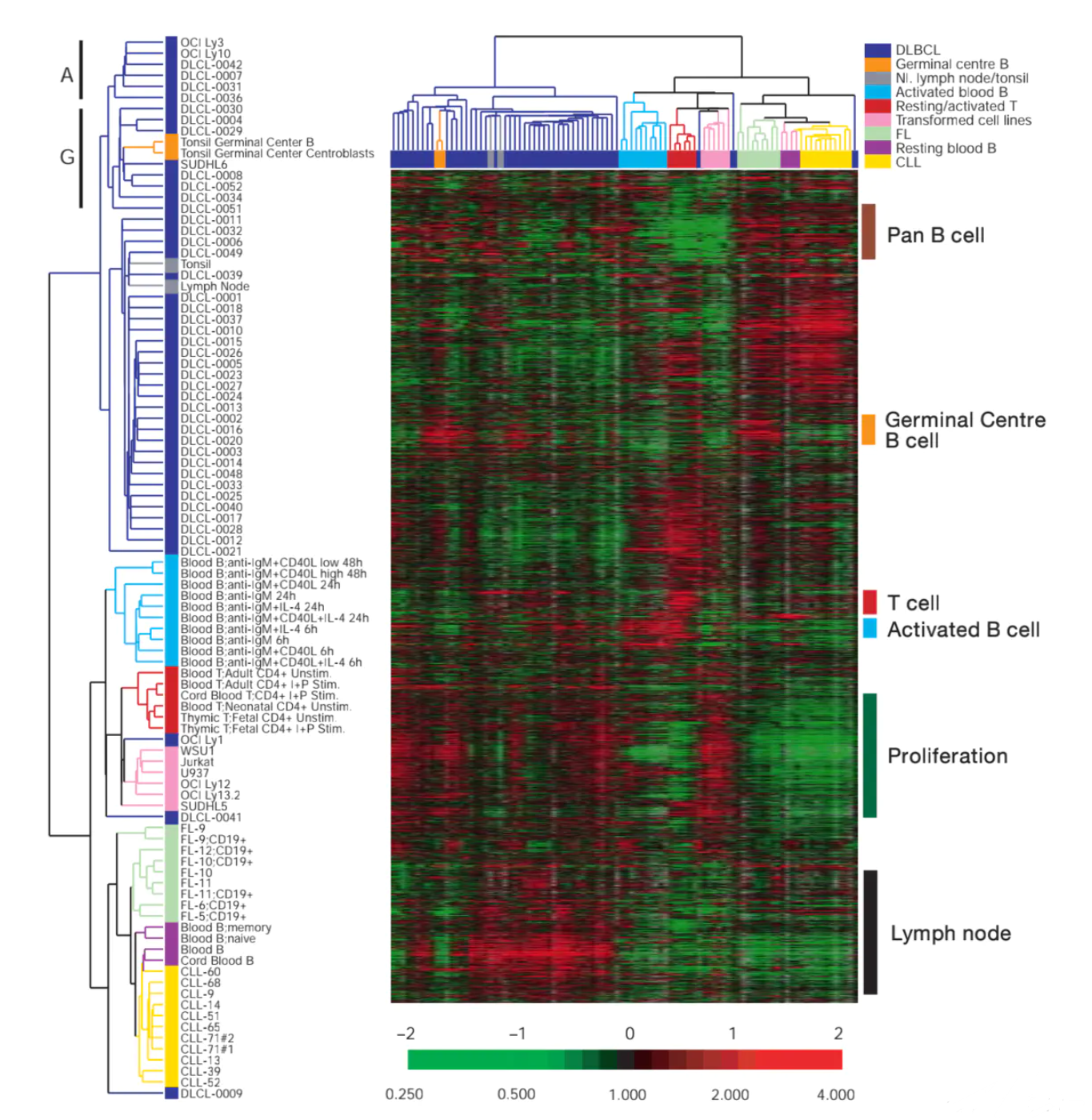
评价
分层聚类的优点是为数据提供了一种直观的表达形式,而且允许算法的使用者根据结果自行决定聚类的深度。分层聚类的缺点是鲁棒性不高:对数据集的些许扰动最终可能会导致整个聚类树状图出现较大的变动。其次,算法的计算复杂度比较高。最后,由于分层聚类的贪心特性,每一次迭代的结果都基于前一次迭代,如果某一次迭代的聚类效果并不好,那么将会影响到之后的所有迭代。
图论聚类
对于一个数据集\(X\),可以建立起一个相似度矩阵\(P\),使得矩阵\(P\)中的每一个元素为对应的两个数据点的相似度:\(P[i,j]=proximity(O_i,O_j)\). 在图论聚类(graph thoertical clustering)中,根据相似度矩阵建立一个相似度图(proximity graph)\(𝒢\),这个图中的每个定点代表一个数据点。两个数据点的连线上根据这两个数据点的相似度被赋予一定权重。如此就将一个聚类问题转化为了图论中的最小割问题(minimum cut)或者最大团(maximum cliques)问题。
最小割问题:
在图论中,去掉其中所有边能使一张网络流图不再连通(即分成两个子图)的边集称为图的割,一张图上最小的割称为最小割。图中所有的割中,边权值和最小的割为最小割。
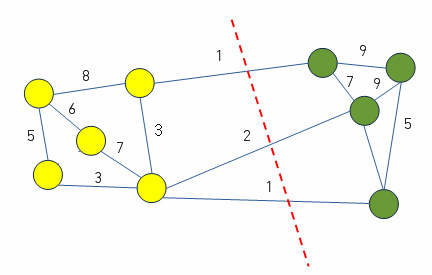
可以理解,在相似度图\(𝒢\)中的最小割代表使得相似度最小的几个连线被断开,形成两个新的簇。
最大团问题:
对于一个无向图\(G=(V,E)\),\(V\)是点集,\(E\)是边集。取\(V\)的一个子集\(U\),若对于\(U\)中任意两个点\(u\)和\(v\),有边\((u,v)∈E\),那么称\(U\)是\(G\)的一个完全子图。如果该完全子图\(U\)不包含在更大的完全子图中,那么称\(U\)是一个团。\(G\)的最大团指的是顶点数最多的一个团。
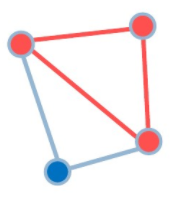
本论文中提到的图论聚类的两种算法为CLICK和CAST。
CLICK算法
CLICK算法(CLuster Identification via Connectivity Kernels)试图将相似度图中具有较大权值连接的顶点聚为一簇。CLICK算法基于一个假设:标准化后的权值服从正态分布。CLICK算法中相似图的某条连接的权重\(w_{ij}\)可以理解为顶点\(i\)和顶点\(j\)被分到同一个簇中的概率。CLICK的迭代过程需要不断地找到整个相似度图\(𝒢\)以及其最小割后的子图的最小割。
之后,CLICK算法有额外的两步对聚类结果进行修饰(refine):
- “采用”步骤(adoption step)用于处理迭代过程之后剩下的单个顶点(singletions),并更新当前的簇。
- “合并”步骤(merge step)用于合并两个相似度超过某个提前设定好的阈值的簇。
CLICK算法的同质性(homogeneity,同一个簇中的个体相似度高)和异质性/分离性(heterogeneity/seperation,不同簇中的个体相似度低,差异大)都比较好。
但是CLICK几乎不能保证迭代过程不走错路,最终导致生成高度不平衡的划分(highly-unbalanced partitions):比如一个将数据集分为数据点数量差异非常悬殊的划分。此外,如果最终的簇与簇之间可能存在交叠的话,CLICK算法也不能处理好这样的数据。对于基因表达数据而言,可以通过经验判断其最终簇和簇之间可能会有交叠,因此CLICK算法可能无法正确处理这样的基因表达数据。
CAST算法
CAST算法认为对数据集正确的簇分类可以表示为一个团图(clique graph)\(ℋ\),这个团图中的每一个团都对应了一个簇。团图的相似度图可以根据以概率\(α\)翻转每条连接得到。对于数据的聚类可以认为是通过对相似度图进行一定的翻转,最终得到一个确定的团图。
CAST算法用亲和度(affinity)来衡量每一个数据点对某个簇的从属关系,并通过一个设定的亲和度阈值(affinity threshold)\(t\)来决定某个数据点是否属于这个簇。在算法的迭代过程中,CAST会不断删除这个簇中亲和度小的数据点,并添加亲和度高的数据点。亲和度阈值实际上是当前这个簇中所有数据点配对的平均相似度,因此无需设置。
CAST算法的优点是不需要用户提前对簇的数量等参数进行设置。但是对于大量数据,寻找亲和度阈值过程的计算量和复杂度都比较高。
基于模型的聚类
基于模型的聚类方法 (Model-based Clustering Methods)的基本其基本思想是:整个数据集的分布一定由有限个已知的概率分布模型叠加而成;其中每一个簇对应了一个已知的概率分布模型。那么整个整个数据集的概率模型可以表示为:
\[L_{mix}(Θ,Γ)=∑_{i=1}^kγ_r^if_i(x_r|θ_i)\] 其中,\(n\)是数据点的数量;\(k\)是簇(即概率模型)的数量;\(x_r\)是一个数据点;\(γ_r^i\)表示\(x_r\)属于簇\(C_i\)的概率,称为隐含参数;\(f_i(x_r|θ_i)\)是引入\(x_r\)后簇\(C_i\)概率密度函数,其中包含一些未知参数\(θ_i\),称为模型参数。
基于模型的聚类方法需要找到使得\(L_{mix}(Θ,Γ)\)最大的所有\(Θ=\{θ_i|1≤i≤k\}\)和\(Γ=\{γ^i_r|1≤i≤k,1≤r≤n\}\)。
通常,可以通过EM算法/期望最大化算法(Expectation-Maximum)找到合适的\(Γ\)和\(Θ\)。EM算法的运行过程分为两步:期望和最大化。
- 首先对每一个簇的概率模型中参数\(Θ\)(比如均值和方差)进行的随机初始化。
- 期望过程(E过程):计算每个数据点\(x_r\)更属于哪一个簇的概率分布,计算每个数据点对最佳簇的\(γ_r^i\),得到\(Γ\)。
- 最大化过程(M过程):利用现在每个簇中的数据点分别对每个簇的概率密度参数\(θ_i\)进行极大似然估计,得到\(Θ\).
然后重新计算每个数据点\(x_r\)更属于哪一个簇的概率分布,如此重复E过程和M过程,直到\(Γ\)和\(Θ\)不再发生变化。
评价
基因表达数据可能会出现单个基因与两个不同的簇有高度相关性的情况。基于模型的聚类的概率性质适合处理这种情况。但是基于模型的聚类假设了每一个簇一定服从某个特定的概率分布模型,事实上这一假设未必符合实际。
基于密度的分层聚类/DHC
基于密度的分层聚类又称为DHC算法(Density-based Hierarchical Clustering),其基本思想是将一个簇视为一个多维的稠密区域。区域核心部分的数据点非常接近,因此具有高密度。这个稠密区域中的数据点之间相互“吸引”(attract):簇中核心区域的数据点会对簇边缘的数据点产生吸引。
DHC将数据集组织为两层结构:第一层使用吸引树(attraction tree)来表示稠密区中数据点之间的关系。树中的每个节点表示稠密区中的一个数据点,节点与节点之间的连线表示两个数据点之间的影响因子(attractor)。具有最高密度的数据点将作为这个树的根部。然而面对较多数据时一层树结构描绘的数据关系较为复杂,因此DHC设计了第二层树结构。在第二层,DHC将第一层的簇结构精炼为密度树(density tree):密度树的每一个节点表示一个稠密区,在密度树中表示为整个密度树的根节点。算法运行最初,整个数据集被视为一个稠密区,然后依据某些规则被划分为多个子稠密区,这些子稠密区表示为根节点的子节点。随着算法的迭代,子稠密区会被不断划分,形成新的子树,直到最终每个稠密区对应一个簇。
评价
DHC可以很有效的从信噪比低的环境中找出共表达基因,因此DHC在噪声环境下的鲁棒性很高。其次,由于DHC一开始就捕捉到了簇的“核心”部分,然后再根据“吸引”的定义划分出簇之间的界限,因此DHC适用于个体高度相关的数据集。但是DHC算法的计算复杂度高,为\(O(n^2)\)。同时,DHC也需要定义什么是数据的“核心”以及数据之间的“吸引”。
总结
基于基因聚类的困难性
基于基因的聚类算法仍然存在许多挑战:
- 通常使用聚类算法的目的是为了揭示出数据本身的性质,因此好的聚类算法应当尽可能少地依赖先验知识,这一点对于基因聚类很难做到。
- 基因表达数据的信噪比非常低,因此对基因表达数据的聚类算法应当对噪声的鲁棒性高。
- 基因表达数据的个体相关性高,聚类结果中的簇之间可能相互重叠。
- 基因表达数据分析不仅要关注哪些基因被分到了同一个簇,也要关注簇与簇之间的关系,并且要关注同一个簇中不同基因之间的关系。聚类算法的结果应当能够用一些方法(比如图)展示这些关系。
评价
使用基于基因的聚类的目的是为了从信噪比低的基因表达数据中找出高度相关的共表达基因个体。传统的聚类方法,例如K均值、SOM以及分层聚类已经被证实在这一领域当中是有用的。然而这些算法在设计之初本来只是为了通用的聚类,并未对基因表达数据做特化处理,因此对于基因表达数据的聚类效果可能效率不高。
而CLICK、CAST、DHC等针对于基因表达数据的聚类算法在实验中已被证实在对基因表达数据的聚类中性能要高于传统的聚类算法。
不过事实上每一种算法的性能都与对应的数据集的性质有关:比如K均值和SOM算法相比于其他算法更加适用于簇数量已知、且只有少数不属于任何簇的数据(outlier)的数据集;对于高信噪比和簇数量未知的数据集,CLICK和CAST的性能较好。
基于样本的聚类
使用基因数据矩阵时,可以发现一些样本组成的表现型往往与某种特定的症状、药物影响有关(比如患有同一种疾病的样本、接受过同一种治疗的样本等等)。基于样本的聚类目的是为了找到表现型的结构(表现型由哪些样本组成)。
生物学经验得知,通常样本的某个表现型只能通过一小部分基因来区分,这些基因的表达水平与类别的区分密切相关。这些基因被称为信息基因(informative gene)。其他的基因由于与这个表现型无关,因此聚类中被视为噪音。通常,基因表达数据的信噪比大约只有0.1,如果直接对数据进行聚类,其结果的可靠性和质量将会大幅度降低。因此基于样本的聚类往往会先筛选出信息基因以降低维度,然后再聚类。
基于样本的聚类主要有两类:监督的分析和非监督的分析。两者的区别在于在筛选和聚类阶段使用的是监督学习还是非监督学习。
基于监督学习筛选的聚类
基于监督学习筛选的聚类假设表现型与样本直接相关,比如样本直接被标注为“患有疾病的”和“未患有疾病的”正负两类。利用这些信息,可以构建一个只包含信息基因的分类器。基于这个分类器,可以对样本进行聚类以匹配它们的表现型,并且可以从表达谱中预测未来的样本标签。分类器的构建过程包括:
- 划分训练集
由于数据集中的样本个数十分有限,训练集通常与原始的数据集中的数据个数具有相同数量级。
- 信息基因选择
信息基因选择可以选择出根据表达模式可以区分具有不同的表现型样本的基因。比如有一个基因在某一类样本中表达水平都比较高,而在其他类样本中表达水平都比较低。信息基因选择的方法包括近邻成分分析以及一些监督学习方法(比如支持向量机和一些基于排序的学习方法)。
- 样本聚类和分类
整个数据集的所有样本依照被筛选出来的信息基因而分类。由于样本数比较小,因此K均值和SOM通常直接被应用进行分类。
非监督的筛选和聚类
基于非监督学习的筛选和聚类方法认为样本没有被标注任何关于表现型的信息。而非监督学习的目的则是自动地发现潜在的表现型信息。
由于没有训练集,相比于监督学习,非监督学习要更加复杂。对于非监督学习,还需要应对如下的两个问题:
- 由于样本数目非常少基因数量非常多,因此数据点在以基因作为维度的高维空间中非常稀疏,传统的聚类方法无法对这样的数据分布进行合理划分。
- 信噪比低:只有非常少的基因是感兴趣的。
对于非监督的筛选和分类,通常有如下的两种策略来解决这些问题:非监督的基因筛选(unsupervised gene selection)和相连聚类(interralated clustering)。
非监督的基因筛选
这种策略中,基因筛选和样本聚类被视为独立的两个过程。首先,基因,也就是特征的维度通过一定方法被减小,然后使用传统的聚类方法对样本进行聚类。
由于没有训练集,选择过程只依赖于统计模型来筛选基因。常用的方法为主成分分析(PCA)法,通过主成分分析可以将基因的维度减少到一定程度,留下的基因即为主成分,称为特征基因。但是利用统计学方法筛选出来的特征基因与生物学角度的信息基因并没有太多重合。
另一种方法是F统计量(F-statistic)筛选出基因表达数据矩阵中具有高方差的基因。这种方法基于的假设是信息基因一定比噪声具有更高的方差——这样的假设对于基因表达数据未必真实。其次,这种方法的有效性基于数据分布。
相连聚类
相连聚类认为基因筛选和样本聚类两个过程彼此是相关联的(interrelated):只要信息基因能够被正确地筛选出来,那么就可以简单地使用传统的聚类方法对样本进行聚类;另一方面,如果样本可以被正确地划分,那么一些基于排序的监督学习方法可以根据基因对划分的相关性来为基因进行排序:与某个划分呈现高相关的基因可以被认为是信息基因。
基于上述理论,相连聚类利用了信息基因和样本划分之间的关系,不断地迭代进行基因筛选和样本聚类。随着算法的不断运行,对样本的划分会不断地接近于真实的划分,最终达到收敛。
Xing和Karp提出了一种基于采样的聚类算法CLIFF,这种算法在每一次迭代中首先会使用一种称为“信息增益排序”(information gain ranking)的算法为某个样本划分\(C\)筛选出与其最相关的基因。接着使用马尔可夫毯(Markov Blanket filter)算法找到这个最相关基因的马尔科夫毯,同时过滤出噪声基因。剩下的基因会被当成特征参与下一轮的样本划分,划分过程使用NCut聚类算法。当两轮样本划分完全相同时,算法达到收敛。
马尔科夫毯(Markov Blanket)
在随机变量的全集\(U\)中,对于给定的变量\(X∈U\)和变量集\(MB⊂U\)且\(X∉MB\),如果有:
\[X⊥\{U-MB-\{X\}\}|MB\] 上式读作“在给定集合\(MB\)(\(|MB\))的条件下,变量\(X\)独立于(\(⊥\))\(U\)与\(MB\)和\(\{X\}\)的补集。”即在全集\(U\)中去掉集合\(MB\)后,变量\(X\)将于\(U\)剩下的部分完全无关。换言之,变量\(X\)通过集合\(MB\)与全集\(U\)中的其他部分建立联系。称\(MB\)是变量\(X\)的马尔科夫毯。
如下图阴影区即为变量\(A\)的马尔科夫毯,去掉阴影区后\(A\)将无法与其他结点建立联系。
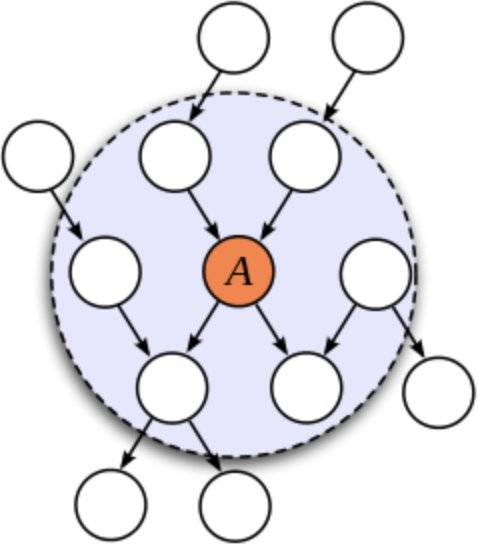
对于\(X\)而言,所有不包含在马尔科夫毯中的变量,即非马尔可夫毯变量都是冗余的。换言之,关于目标变量\(X\)的所有信息都包含在\(X\)的马尔可夫毯中。如果需要要了解目标变量的分布情况,仅需要了解其马尔可夫毯的信息即可,而不需要对于整个数据集进行了解。
马尔可夫毯是进行特征冗余性分析的一种常用的工具。在一个特征空间中,目标特征的马尔可夫毯包含了它的所有信息。目标特征的详细信息可以从它的马尔可夫毯中获得,非马尔可夫毯可以看成是目标特征的冗余特征。因此,通过发现目标特征的马尔可夫毯,就可以准确地确定目标特征的冗余特征,从而降低特征空间的维数。
CLIFF算法的问题是NCut算法对噪声和不属于任何划分的样本非常敏感,因此整个算法的鲁棒性较低。
总结
对于基于样本的聚类,最需要解决的问题就是低信噪比的问题。为此,基于监督学习筛选的聚类使用样本中标注的表现型信息进行学习然后筛选出信息基因,这种方法被广泛应用而且比较简单,而且由于大部分的样本都被划分为训练集,因此比较容易能够达到高准确度。
非监督的筛选和聚类更为复杂,非监督的基因筛选和相连聚类用于解决低信噪比问题:非监督的基因筛选由于基于统计学模型,因此性能和有效性高度依赖于数据集的统计分布。相连聚类考虑了基因筛选和聚类之间的关系,但是由于每次迭代中的基因筛选都是不可逆的,因此每次聚类的结果都是局部最优而非全局最优。
基于样本聚类的问题
- 簇的数量\(K\)的确定。
通常表现型的数量非常的少并且已知,因此对于基于样本的聚类,\(K\)通常是已知的。
- 时间复杂度
由于样本数量\(m\)远远小于基因数量\(n\),因此只关心对基因数量的时间复杂度。不管是监督学习还是非监督学习,其中的每一个基因只会被遍历一次,因此两种算法的时间复杂度通常是\(O(n)\). 而相连聚类的时间复杂度为\(O(n,l)\)其中\(l\)为算法迭代的次数,在算法开始运行前\(l\)难以被估计。
子空间聚类
前面介绍的两种聚类方法都是“全局聚类”(global clustering):对于给定的数据集,特征空间被全局确定且被所有的簇共享,最终所有的簇彼此不会重叠。然而,分子生物学的常识认为一个细胞过程只有非常少数的样本参与,同时一个基因可能会参与多个细胞过程,在某些情况下,有些基因甚至根本不参与任何细胞过程。
子空间聚类(subspace clustering)可以找到特征空间中的某些子空间中的簇。最终由子空间聚类找到的子空间簇可能重叠,也有可能一些个体不属于任何子空间簇——这样的特性适合于基因表达数据的特点。
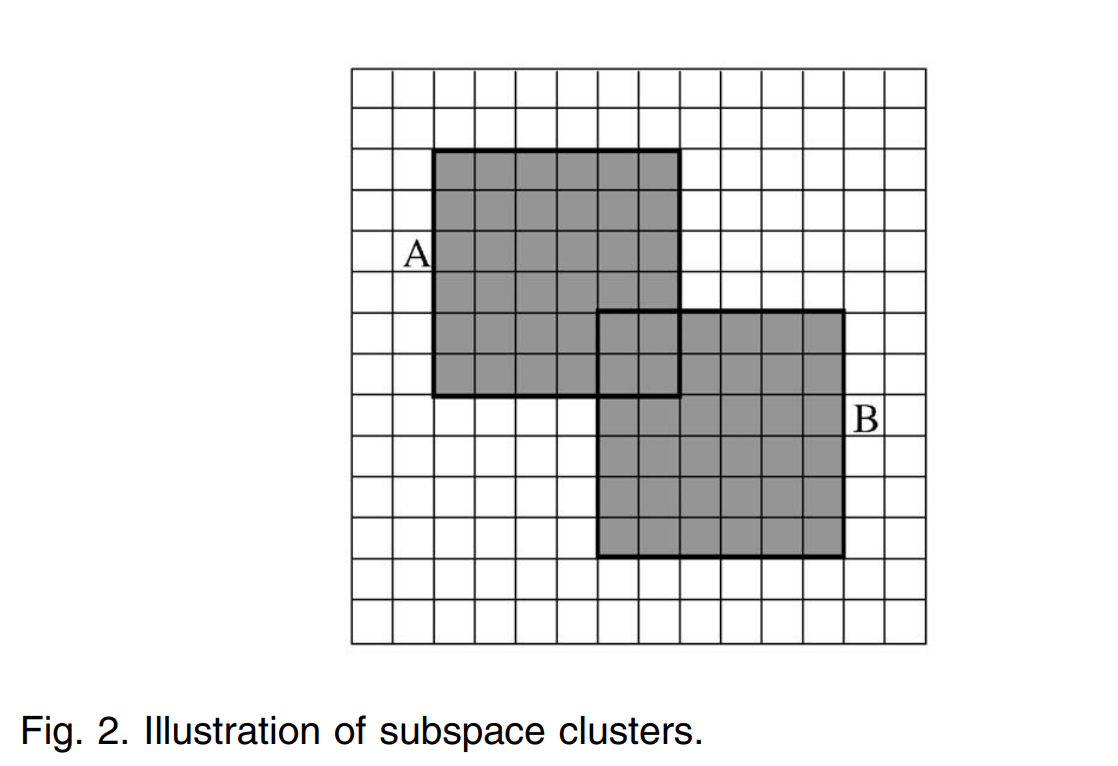
定义基因表达数据矩阵中的一个子矩阵称为一个块(block),它是基因表达数据的特征空间中的一个子空间。毫无疑问,在整个特征空间中找到所有的子空间是一个NP-hard问题。因此,子空间聚类通常会首先定义块,然后通过某些为基因表达数据特化的启发式算法来进行聚类。通常这些聚类算法同等地将基因和样本视为特征或者数据点。
耦合双向聚类
耦合双向聚类(CTWC,Coupled two-way clustering)将一个块视为含有特征子集\(ℱ_i\)和数据子集\(𝒪_j\)的“稳定”簇(stable cluster),其中特征点和数据点既可以是基因也可以是样本。“稳定”簇的含义是:只有\(ℱ_i\)中的特征被用于\(𝒪_j\)的聚类。且\(𝒪_j\)低于某一阈值时不会被划分。此外,CTWC使用了一种启发式的方法来避免遍历:只有前一个迭代中的稳定簇才能进入下一个迭代。
CTWC开始于一个包含所有基因\(G_0\)和所有的样本\(S_0\)的特征空间\((G_0,S_0)\),接着对\(G_0\)和\(S_0\)使用一种分层聚类算法(SPC, superparamagnetic clustering algorithm)找到基因和样本的稳定簇\(G_1^i\)和\(O_1^i\).
CTWC动态地记录下两个分别包含基因稳定簇和样本稳定簇的列表:\(GL\)和\(SL\)。在每一次迭代中,之前从未配对过的一个基因子集和一个样本子集从\(GL\)和\(SL\)被随机选择出,随机配对组成子空间,然后聚类并找到稳定簇,将新的稳定簇加入\(GL\)和\(SL\)。每次配对都会被指针记录下来。
当没有新的稳定簇生成时,算法收敛。
评价
CTWC的聚类结果会随着初始选择的块的不同而不同,因此对起始点敏感。此外,CTWC的聚类结果中有非常多的冗余,并且难以解释。
Plaid 模型聚类
Plaid模型将基因表达数据视为多个数据层的和,其中每一个数据层代表了一种只包含一部分基因和样本参与的特定生物过程。Plaid模型表示为:
\[Y_{ij}=∑_{k=0}^Kθ_{ijk}ρ_{ik}𝒦_{jk}\] 表示基因\(i\)在样本\(j\)中的表达水平\(Y_{ij}\)。
其中\(θ_{ij0}\)表示整个数据集的整体的表达水平,\(θ_{ijk}\)表示第\(k\)层的生物过程中的表达水平对整体表达水平的贡献;\(ρ_{ik}\)代表基因\(i\)是否参与第\(k\)层的生物过程:如果参加,\(ρ_{ik}=1\);如果不参加,\(ρ_{ik}=0\);\(𝒦_{jk}\)代表样本\(j\)是否参与第\(k\)层的生物过程:如果参加,\(𝒦_{jk}=1\);如果不参加,\(𝒦_{jk}=0\).
整个聚类过程遍历每一层的生物过程,使用EM算法来预测模型参数。具体而言,假设前\(K-1\)层已经被遍历,算法在第\(K\)层需要最小化如下的方差和:
\[Q=\frac{1}{2}\sum_{i=1}^n\sum_{j=1}^m(Z_{ij}-θ_{ijk}ρ_{ik}𝒦_{jk})^2\] 其中,
\[Z_{ij}=Y_{ij}-θ_{ij0}-∑_{k=0}^{K-1}θ_{ijk}ρ_{ik}𝒦_{jk}\] 为前\(K-1\)层的残差。
当\(Q\)小于某个阈值时,算法停止。
评价
Plaid模型聚类在酵母细胞的基因表达数据实验中被证明是有效的。然而,Plaid模型本身基于一个不可信的假设:如果一个基因参与了多个细胞过程,那么其总体的表达水平为其在每个细胞过程的表达水平之和。
双聚类
双聚类算法可以同时聚类矩阵的行列。双聚类算法中使用均方残差(mean-squared residue)来衡量块中基因与条件之间的相关性。记\(G'\)和\(S'\)分别为基因和样本的一个子集,有\((G',S')\)的均方残差为:
\[H(G',S')=\frac{1}{|G'||S'|}∑_{i∈G',j∈S'}(w_{ij}-η_{iS'}-η_{G'j}+η_{S'G'})^2\] 其中\(η_{iS'}=\frac{1}{|S'|}∑_{j∈J}w_{ij}\),\(η_{G'j}=\frac{1}{|G'|}∑_{i∈I}w_{ij}\),即基因表达数据矩阵中某个块的行、列的均值。
\(η_{S'G'}=\frac{1}{|G'||S'|}∑_{i∈G',j∈S'}w_{ij}\)为块的均值。
低的均方残差代表这个块的方差高于正常值,更易于识别。因此定义某个均方残差小于等于某个阈值\(δ,H(G',S')≤δ,δ>0\)的块为双向簇(bicluster)/δ簇(δ-cluster,在δ聚类中)。
找到双向簇的方法是每次增加或者减少块中的一行或者一列,重新计算\(H\),应用可以使\(H\)减少最快的行列增减。最终\(H\)不发生变化或者\(H(G',S')≤δ\),那么当前的子矩阵即为一个双向簇。
找到一个双向簇之后,这个双向簇对应的所有的元素被替换为随机数。然后再次原矩阵中寻找双向簇,直到满足设定数量的双向簇被找到。
然而双聚类算法不能保证先找到的双向簇的效果比后找到的双向簇效果更好,这增加了对聚类结果的解读。其次,双聚类使用随机数掩盖原来的双向簇,使得无法发现与原来双向簇重叠的新的双向簇。
δ聚类
一种改进双聚类算法的方法是δ聚类算法(δ-cluster)。这种算法使用平均残差(average residue)来衡量相关性。此外,算法每次同时对随机选择的\(K\)个子矩阵通过增减子矩阵的行列来查找δ簇。这样一定程度上避免了无法查找到相互重叠的双向簇的问题。同时平均残差只通过存在的值来计算得出,对缺失数值的鲁棒性更强。但是δ聚类的时间复杂度更高,为\(O((n+m)×n×m×l×k)\)。\(k\)是最终找到的δ簇的个数,\(l\)是迭代次数。
总结
子空间聚类对基因表达数据矩阵中的一些块进行聚类。不同的子空间聚类方法采用了不同的贪心启发式算法来近似最优解。子空间聚类的一个优点是其聚类过程是可追溯的。
聚类效果验证
不同的聚类方法有不同的聚类参数,在聚类算法完成之后,有必要通过某种途径来统一各种聚类算法的性能指标,以便选择出最适合的聚类算法。聚类验证(cluster validation)就是衡量不同聚类算法的可靠性和质量的过程。进一步地,聚类验证从三个方面衡量某个聚类算法:
- 聚类的质量
指聚类完成后,对不同簇之间的差异和同一个簇内部的数据点相似度的衡量,质量好的聚类算法在聚类完成后,不同簇之间的差异应当大,同一个簇内部的差异应当小。
- 聚类的合理性
指聚类结果应当符合先验知识和常识认知。
- 聚类的可靠性
指聚类结果并非偶然形成的概率。
聚类的质量
同质性
简单来说,同质性(homogeneity)衡量了某个簇\(C\)中数据点之间的相似程度。对于同质性的定义有非常多种,比如:
\[H(C)=\frac{∑_{i,j∈C,i≠j}similarity(O_i,O_j)}{||C||(||C||-1)}\] 这种方法衡量了\(C\)中每一种可能的数据点两两配对之间的相似度,并进行了归一化处理,得到平均相似度。
另一种可行的方法是衡量\(C\)中每一种可能的数据点与\(C\)的质心(centroid)\(\overline{O}\)的平均相似度:
\[H(C)=\frac{∑_{i∈C}similarity(O_i,\overline{O})}{||C||}\] 在某些条件下,使用\(C\)最大或者最小的相似度也能够有好的效果。
整个聚类结果的同质性可以定义为:
\[H_{avg}=\frac{1}{N}∑||C_i||H(C_i)\]
分离性
分离性(separation)衡量了两个簇\(C_1\)和\(C_2\)之间的差异性,比如如下的定义:
\[S(C_1,C_2)=\frac{∑_{i∈C_1,j∈C_2}similarity(O_i,O_j)}{||C||(||C||-1)}\] 整个聚类结果的分离性可以定义为:
\[S_{avg}=\frac{1}{∑_{C_i≠C_j}||C_i||||C_j||}∑_{C_i≠C_j}(||C_i||||C_j||)S(C_iC_j)\]
可以发现,如果同一个簇中两个数据点相似度越高,不同簇的两个数据点相似度越低,那么算法的同质性越大、分离性越小,算法的质量越高。
聚类的合理性
聚类的合理性可以通过对比聚类结果\(𝒞=\{C_1,...,C_p\}\)和常识\(P=\{P_1,...,P_s\}\)得到。二者都可以转化为矩阵,对于聚类结果\(𝒞=\{C_1,...,C_p\}\),如果\(O_i\)与\(O_j\)位于同一个簇,那么矩阵中第\(i\)行\(j\)列的元素\(C_{ij}=1\),反之为0;对于常识\(P\)也可以通过相同的方法得到一个二进制矩阵,对比两个矩阵。定义\(n_{C,P}\)中的下标分别代表数据对\((O_i,O_j)\)在矩阵\(C\)、矩阵\(P\)中的结果,其本身表示这种结果的数据对个数。(比如\(n_{1,1}\)表示数据对\((O_i,O_j)\)在矩阵\(C\)中\(C_{ij}=1\)、矩阵\(P\)中\(P_{ij}=1\)的数据对个数)。 有如下指标可以衡量聚类的合理性:
- 兰德系数(Rand index)
\[Rand=\frac{n_{11}+n_{00}}{n_{11}+n_{01}+n_{10}+n_{00}}\] 即聚类结果完全符合常识的比例。
- 杰卡德系数(Jaccard coefficient)
\[J=\frac{n_{11}}{n_{11}+n_{00}n_{11}+n_{01}+n_{10}}\] 表示\(C\)与\(P\)的交集占\(C\)与\(P\)并集的比例。即聚类结果与常识重合的部分占聚类与常识合并的比例。
- 闵可夫斯基度量(Minkowski measure)
\[M=\sqrt{\frac{n_{01}+n_{10}}{n_{11}+n_{01}}}\] 表示\(C\)与\(P\)不同的部分占常识的比例。即聚类结果与常识不同的部分占常识的比例。
对于基因表达数据,杰卡德系数和闵可夫斯基度量可能更加适合,因为算法的运行结果\(n_{00}\)会远大于其他三者,导致兰德系数不能真实反映聚类的合理性。
聚类的可靠性
聚类的可靠性指聚类结果并非偶然形成的概率。有如下的两种方法来衡量聚类的可靠性。
假设检验和P值
假设检验中需要做出两个完全对立的假设:零假设/原假设和备择假设。如果零假设为真,且在一次抽样中发生了小概率事件,那么有理由拒绝零假设的真实性,从而接受备择假设。
P值(P-value)是假设检验中假设零假设为真时观测到至少与实际观测样本相同极端的样本的概率。很小的P值说明在零假设下观测极端结果的发生概率很小。在实验中一般选取P值大于0.05(称为显著性水平)作为结论是否可信的阈值,如果P值小于0.05,那么说明结论是可信的。
在基因表达数据中使用了超几何分布来衡量在有\(n\)个基因的簇中观察到至少\(k\)个功能基因(参与某些功能的基因)的概率:
\[P=1-∑_{i=0}^{k-1}\frac{C_f^i C_{g-f}^{n-i}}{C_g^n}\] 其中\(f\)是功能基因的总数;\(g\)是基因组中所有基因的数量;
在基因表达数据中这一概率小于0.05%时,表明聚类结果是可信的。
预测强度
预测强度基于的理论是:如果聚类结果可以反应真实的聚类结构,那么基于这个聚类结果的预测器应当可以正确预测出新样本所属的簇。
由于基因表达数据的样本量太少,因此往往采用交叉验证的方法。
展望
正如文中中提出的那样,现在有非常多的聚类算法可以被应用于基因表达数据。然而,如今面临的问题是对特定的基因表达数据集应当如何选择最合适的聚类方法。然而,没有任何一个聚类算法在基因表达数据处理的任何一个细分领域被认为是绝对的胜者。通常研究人员仍然需要选择一系列的聚类算法进行结果对比和测试。本文中给出了三个方面的算法验证方法,但是现如今还没有统一的有效性度量。不仅如此,各种聚类算法的性能在很大程度上仍然与不同数据集本身的特征分布有关。因此聚类算法的测试和验证通常会考虑多个度量。
另一个问题是,现存的大部分聚类算法无法对不同的聚类粒度要求进行适配。对于基因表达数据,本文作者建议最好避免直接对数据集进行划分,转而使用图像表示出聚类结构,由算法的运行者根据需要选择聚类粒度。一些聚类算法比如SOM和分层聚类可以实现对聚类结构的图像表示,但是这些算法可能不能满足不同用户对子数据集的粒度要求。
聚类被普遍认为是一个 “无监督”的学习问题。在进行聚类之前,关于数据集的 “全局”信息,如聚类的总数和对象空间中的完整数据分布,通常是未知的。然而,关于基因表达数据集的一些先验知识往往是可用的。例如,一些基因的功能已经在文献中得到研究,这可以为聚类提供指导。此外,如果已知一些实验条件的组别是强相关的,这些不同组别下的聚类结构的差异可能是特别值得关注的。如果聚类算法在执行聚类任务时能将这种部分知识整合为一些聚类约束条件。
可以预期,聚类结果将更具有生物学意义。这样一来,聚类就不再是是一个 “纯粹的”无监督过程,而成为对数据集的互动探索。